
Abstract
BACKGROUND AND PURPOSE: Supratentorial primitive neuroectodermal tumors and pineoblastomas have traditionally been grouped together for treatment purposes. Molecular profiling of these tumors has revealed a number of distinct entities and has led to the term “CNS-primitive neuroectodermal tumors” being removed from the 2016 World Health Organization classification. The purpose of this study was to describe the MR imaging findings of histologically diagnosed primitive neuroectodermal tumors and pineoblastomas and correlate them with molecular diagnoses and outcomes.
MATERIALS AND METHODS: Histologically diagnosed primitive neuroectodermal tumors and pineoblastomas were enrolled in this Children's Oncology Group Phase III trial, and molecular classification was retrospectively completed using DNA methylation profiling. MR imaging features were systematically studied and correlated with molecular diagnoses and survival.
RESULTS: Of the 85 patients enrolled, 56 met the inclusion criteria, in whom 28 tumors were in pineal and 28 in nonpineal locations. Methylation profiling revealed a variety of diagnoses, including pineoblastomas (n = 27), high-grade gliomas (n = 17), embryonal tumors (n = 7), atypical teratoid/rhabdoid tumors (n = 3), and ependymomas (n = 2). Thus, 39% overall and 71% of nonpineal tumor diagnoses were discrepant with histopathology. Tumor location, size, margins, and edema were predictors of embryonal-versus-nonembryonal tumors. Larger size and ill-defined margins correlated with poor event-free survival, while metastatic disease by MR imaging did not.
CONCLUSIONS: In nonpineal locations, only a minority of histologically diagnosed primitive neuroectodermal tumors are embryonal tumors; therefore, high-grade glioma or ependymoma should be high on the radiographic differential. An understanding of molecularly defined tumor entities and their relative frequencies and locations will help the radiologist make more accurate predictions of the tumor types.
ABBREVIATIONS:
- ATRT
- atypical teratoid/rhabdoid tumor
- EP
- ependymoma
- ET
- embryonal tumor
- ETMR
- embryonal tumor with multilayered rosettes
- GBM
- glioblastoma multiforme
- HGG
- high-grade glioma
- HGNET
- high-grade neuroepithelial tumor
- MB
- medulloblastoma
- NOS
- not otherwise specified
- PBL
- pineoblastoma
- PNET
- primitive neuroectodermal tumor
Historically, supratentorial primitive neuroectodermal tumor of the central nervous system (CNS–PNET) and pineoblastoma (PBL) have been considered embryonal tumors (ETs) histopathologically, similar to medulloblastomas (MBs), though the classification has been a topic of much debate.1,2 CNS-PNET and PBL have thus been treated as a single group using protocols designed for high-risk medulloblastomas.3,4 In recent years, molecular profiling using genome-wide DNA methylation of histopathologically diagnosed CNS-PNETs has revealed a wide spectrum of distinct molecular entities, including high-grade gliomas (HGGs), atypical teratoid/rhabdoid tumors (ATRTs), ependymomas (EPs), and at least 4 new molecular entities.5⇓–7 The 2016 World Health Organization classification has removed CNS-PNET as a diagnostic category, in part substituting a broad group termed “CNS embryonal tumors, not otherwise specified” (NOS), in addition to more specific entities, such as embryonal tumor with multilayered rosettes (ETMR).8
Molecularly defined entities may more accurately predict clinical outcomes when compared with standard histopathologic diagnoses in CNS-PNET,7 as well as in other pediatric CNS tumors such as medulloblastoma and ependymoma.9,10 However, practical barriers such as cost, availability, timeliness of results, and assay certification can hamper use. In recent years, there has been growing interest in correlating imaging features with molecular markers in an attempt to identify imaging phenotypes that may serve as surrogates for molecular subtypes.11,12 This radiogenomic approach has been applied with some success in CNS tumors such as glioblastomas,13,14 medulloblastomas,15 ATRT,16 and non-CNS tumors.17
The Children's Oncology Group study, ACNS0332, a multicenter Phase III prospective trial, investigated 2 approaches for treatment intensification, the addition of carboplatin during irradiation and the addition of adjuvant isotretinoin, in patients diagnosed with either CNS-PNET/PBL or high-risk medulloblastoma in 2 parallel randomized strata. The results from the completed CNS-PNET/PBL portion of the trial, including an analysis of molecular profiles and patient outcomes, have been published.7 These results have shown that the molecularly diagnosed HGG had significantly worse survival compared with supratentorial embryonal tumors and PBL.7 Thus, the distinction between these 2 categories is critical to the management of these patients.
As a part of this trial, MR imaging of the brain and spine was performed at multiple time points and submitted for central review. The current report focuses on the MR imaging features of CNS-PNET/PBL and their correlation with molecular subtypes and outcomes. Identifying reliable correlations would facilitate imaging-guided clinical decision-making when molecular profiling is unavailable or delayed.
MATERIALS AND METHODS
Patient Cohort
For the strata included in this report, children from 3 to 22 years were eligible, who had newly diagnosed primary CNS-PNET or PBL by institutional pathologists per the 2007 World Health Organization classification system, defined as undifferentiated or poorly differentiated tumors with the capacity for divergent differentiation. Subjects had minimum functional scores (Karnofsky/Lansky) of 30 and adequate renal, marrow, and hepatic function and were staged with spinal CSF cytology and MR imaging of the brain and spine. Institutional review board approval and individual informed consent were obtained before enrollment, and the study was registered with clinicaltrials.gov (NCT00392327).
Molecular Analysis
DNA methylation profiling was performed for all cases with sufficient tumor DNA using the Infinium HumanMethylation450 (450k; https://www.illumina.com/products/by-type/microarray-kits/infinium-methylation-epic.html) or the EPIC BeadChip arrays (http://emea.support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html), and the tumors were classified using the recently developed brain diagnostic classifier algorithm (www.molecularneuropathology.org).18 The methods are described in more detail in earlier publications.5,7,19
Neuroimaging Guidelines and Central Review
MRIs of the brain without and with contrast were obtained at diagnosis and after definitive surgery (within 72 hours). For patients who underwent stereotactic biopsy only, a postoperative MR imaging was not required. MR imaging of the spine with contrast was obtained within 10 days of surgery if done preoperatively, and within 28 days of surgery if done postoperatively. Additional MRIs of the brain and spine were obtained at the end of radiation therapy, the end of maintenance chemotherapy, at relapse (if any), and at best response.
For this multi-institutional study, guidelines were provided for the technical parameters of MR imaging. The minimum recommended sequences for the brain included sagittal and axial T1-weighted, axial T2-weighted, axial T2 FLAIR, axial diffusion-weighted, and postcontrast axial and sagittal T1-weighted. The minimum recommended sequences for the spine included postcontrast sagittal and axial T1-weighted imaging. The studies were performed on a variety of scanners from different vendors, including both 1.5 and 3T field strengths, and with varying technical parameters.
The MR imaging studies were retrospectively reviewed after treatment completion by 2 experienced pediatric neuroradiologists by consensus (L.A.H. and A.J.), blinded to the histopathologic and molecular diagnoses. Only the MR imaging studies deemed technically acceptable by the central reviewers were included in this analysis, and there was no opportunity to obtain repeat or additional imaging because of the retrospective nature of the review. MR imaging features of the lesions were recorded including location, laterality, size, margins, surrounding edema, enhancement, cyst/necrosis, hemorrhage/calcification, and metastasis. The size was measured as the longest linear dimension in centimeters. The margins were described as well-defined or ill-defined (>50% margins were indistinct). The surrounding edema was assessed as absent, <2 cm from the tumor margin, or >2 cm from the tumor margin. The extent of enhancement of the solid portion of the tumor was categorized subjectively as none, <25%, 25%–75%, and >75%. The degree of enhancement was subjectively assessed as none, mild, moderate, or marked. The presence of cysts or necrosis and low signal on T2 or gradient recalled-echo sequences suspicious for calcification or hemorrhage was noted, and both of these findings were subjectively quantified as involving <25%, 25%–50%, or >50% of the tumor.
The radiographic presence of intracranial and spinal metastasis was assessed and, in conjunction with CSF cytology, was used to assign the M stage (modified Chang staging). Also, the postoperative MRIs were reviewed, and the extent of resection was classified as biopsy (<10%), partial (10%–49%), subtotal (50%–95%), radical subtotal (>95%), and gross total (no visible tumor on imaging).
Outcome Analysis
“Event-free survival” was defined as the time interval from the date of study enrollment to the date of first event (disease progression or recurrence, second malignant neoplasm, or death from any cause) or to the date of last follow-up for patients without events.
Statistical Analysis
The exact Wilcoxon rank sum test was used to compare continuous variables among patient groups. The Fisher exact test and the exact χ2 test were used to compare distributions of categoric variables. Outcome estimates were obtained using the method of Kaplan and Meier. The log-rank test was used to compare outcome distributions. Cox regression was used to examine tumor size as a continuous predictor of outcome. Two-sided P values are reported. Note that patient numbers were quite small in some patient groups. Data frozen as of December 31, 2016, were used for this analysis.
RESULTS
Between March 2007 and August 2014, eighty-five patients with institutionally diagnosed CNS-PNET/PBL were enrolled and randomized among the 4 trial regimens. Of these, 56 patients met the inclusion criteria for the current analysis, including availability of molecular classification by DNA methylation and complete imaging datasets.
Methylation profiling revealed a spectrum of molecular diagnoses broader than those found by histopathology (see Table 1 for further elaboration of these diagnoses) and included PBL (n = 27); HGG (n = 17, including 8 glioblastoma multiforme [GBM]_G34s, 5 GBM_MYCNs, 2 DMG_K27s, 2 GBM_MIDs); ATRT (n = 3, including 2 ATRT_SHHs, 1 ATRT_MYC); CNS neuroblastoma with FOXR2 activation (n = 3); ependymoma with positive RELA fusion (EP-RELA, n = 2); medulloblastoma with Wnt activation (MB_WNT, n = 1); ETMR (n = 1); and high-grade neuroepithelial tumor with MN1 alteration (HGNET-MN1, n = 1). One tumor could not be classified and was designated as embryonal tumor, NOS (ET NOS, n = 1). The molecularly diagnosed supratentorial embryonal tumors were grouped together with PBLs, and the combined group is referred to as PBL/ET hereafter (n = 37, including the following: 27 PBLs, 3 CNS neuroblastomas, 3 ATRTs, 1 MB, 1 ETMR, 1 HGNET, and 1 ET NOS) (Table 1). The rest of the subgroups, including HGG and EP, neither of which were intended for inclusion in the trial, were combined into 1 group, hereafter referred to as nonembryonal tumors (non-ET; n = 19, including: 17 HGGs and 2 EPs). Of note, ATRT, although an embryonal tumor, has historically been considered a unique subset with specific treatment algorithms and was also not intended for trial inclusion. However, keeping with the embryonal tumor definition, we included these under PBL/ET.
Molecular diagnoses for tumors with both imaging and methylation profiles available (n = 56)a
For the included 56 patients, the median age at diagnosis was 9 years (range, 3–18 years). The median age for patients with molecularly diagnosed PBL/ET was 8.6 years (range, 3–18 years), and that for patients with non-ET was 11.0 years (range, 3.8–16.1 years), with no significant difference (P = .21). There were 22 males (39%) and 34 females (61%) overall. In the PBL/ET group, there were 11 males (30%) and 26 females (70%), while in the non-ET group, there were 11 males (58%) and 8 females (42%). The median age at diagnosis for pineoblastomas in our study was 8.7 years (range, 3–18 years).
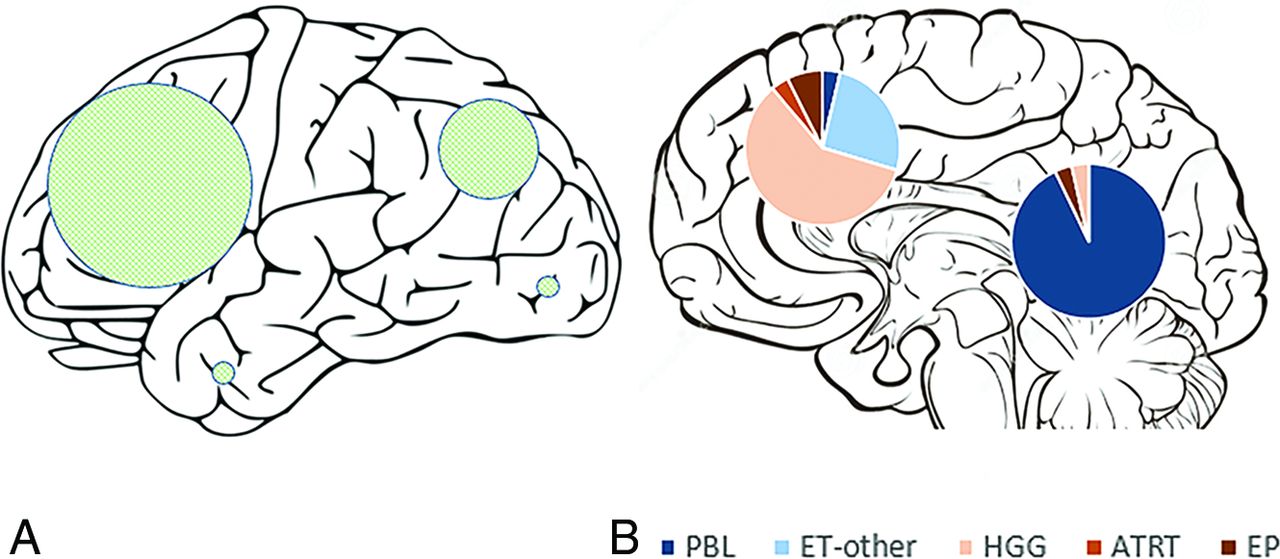
Overall, 28 tumors involved the pineal region and 28 were extrapineal (Table 2 and Fig 1). Among the extrapineal tumors, 64% (18/28) belonged to the non-ET group, compared with only 7% (2/28) of the pineal region tumors (P < .001). Twenty-four of the extrapineal tumors were centered in the parenchyma, and 4, within the ventricles. For the parenchymal tumors, frontal lobe involvement was most common (56%), followed by parietal lobe involvement (36%), with temporal and occipital lobe involvement being rare (7% each) (Fig 1).
Molecular diagnoses for tumors by pineal and nonpineal locations

Schematic representation of the external surface of cerebral hemispheres (A) shows the locations of hemispheric tumors, with the sizes of the shaded circles proportional to the number of tumors in each lobe. A midline sagittal section of the brain (B) shows pineal and nonpineal tumors, with pie diagrams representing the subgroups.
Considering the locations of individual tumor categories, almost all (26 of 27) pineoblastomas were centered in the pineal cistern, with variable involvement of the third ventricle. In 1 patient, the tumor was centered more anteriorly in the third ventricle, without pineal cistern involvement. Five PBLs had tail-like extensions into the cerebral aqueduct, and PBLs rarely demonstrated parenchymal invasion. There were 3 ATRTs, of which 2 were centered in the cerebral hemispheres, and 1, in the pineal cistern. Of the remaining 7 embryonal tumors, 5 were centered in the cerebral hemispheres and 2 were located within the lateral ventricles (Fig 1). The hemispheric embryonal tumors included 3 CNS_NBs, 1 HGNET_MN1, and 1 ET, NOS, while the 2 lateral ventricular embryonal tumors included 1 each of ETMR and MB_WNT subtypes.
In the HGG subgroup, 16 of 17 tumors primarily involved the cerebral hemispheres, and one was centered in the lateral ventricle. Both of the molecularly diagnosed EPs were centered in the cerebral hemispheres.
MR imaging features were compared between the 2 broad groups: PBL/ET and non-ET (Table 3). The median tumor size for the non-ET group was 6.2 cm (range, 2.7–9.3 cm), while that for PBL/ET group was 3.6 cm (range 1.1–9.1 cm), and the difference was statistically significant (P < .001). Thirty-two percent of tumors in the non-ET group (6/19) had ill-defined margins, compared with none in the PBL/ET group (P < .001). Of note, all 6 tumors with ill-defined margins were HGG. Perilesional edema was seen in 74% of the non-ET group (14/19) compared with 14% in the PBL/ET group (5/37), also statistically significant (P < .001). Enhancement was seen in nearly all tumors in both non-ET (19/19) and PBL/ET (36/37) groups, though 47% of tumors in the non-ET group had >75% enhancement, compared with 73% of tumors in the PBL/ET group (P = .08), suggesting a tendency for more homogeneous enhancement in PBL/ET. Most tumors in both groups demonstrated restricted diffusion, cysts/necrosis, and calcification/hemorrhage, with no significant differences for any of these parameters (Table 3).
MR imaging features by tumor group (all patients n = 56)
None of the patients with non-ET had intracranial or intraspinal metastasis identified by MR imaging, while 32% of those with PBL/ET (12/37) had metastasis by MR imaging (1 intracranial, 5 intraspinal, and 6 both) (Table 3). One patient in each of the non-ET and PBL/ET groups had CSF cytology positive for malignant cells, but no detectable metastasis by MR imaging.
After excluding PBL, statistical associations were examined between the remaining ET (n = 10) and non-ET (n = 19) groups. The median age at diagnosis for this ET subgroup was slightly lower (8.4 years) compared with the non-ET group (11 years), though the difference was not statistically significant (P = .10). None of the studied MR imaging parameters showed any statistically significant differences, though we observed some trends (Table 4). Ill-defined margins were observed in 31.6% of the non-ET compared with none in the ET group. Calcification and hemorrhage were more commonly seen in the non-ET (73.7%) compared with ET (40%) group.
MR imaging features by tumor group after excluding pineoblastomas (n = 29)
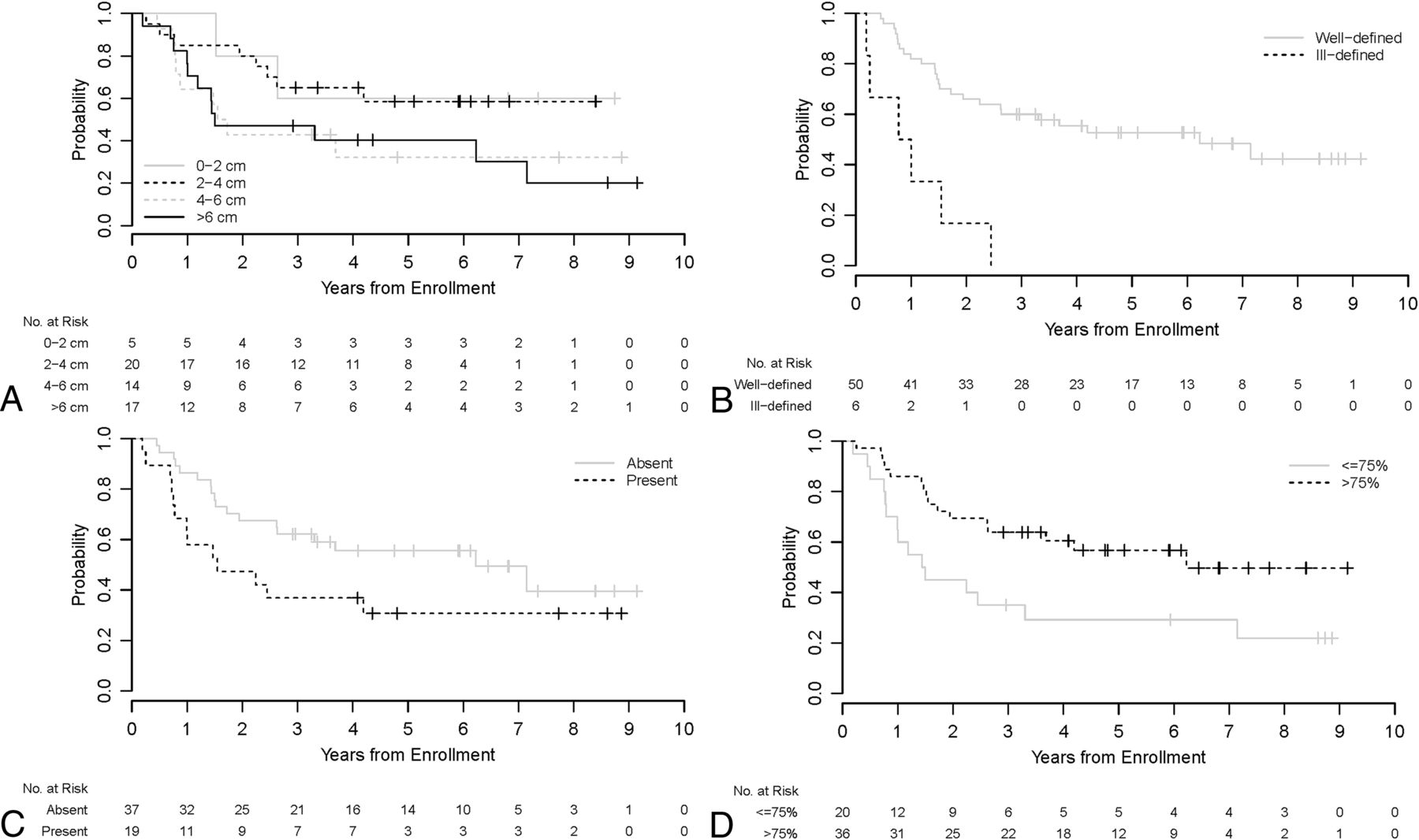
A univariable analysis of imaging parameters with event-free survival was performed (Fig 2). Larger tumor size and ill-defined margins were statistically significant predicators of worse outcomes (P = .023 and .006, respectively).

Kaplan-Meier curves show event-free survival distributions by tumor size (A), margins (B), presence of edema (C), and percentage enhancement (D) for all patients (n = 56).
DISCUSSION
To our knowledge, this is the largest series describing the MR imaging findings of histologically diagnosed supratentorial PNET and PBL and the first to correlate MR imaging features with molecular diagnoses and patient outcomes.
The rapid advances in understanding of the genomic characteristics of tumor cells have led to reassessment of tumor classification and traditional risk factors.19⇓–21 In particular, both supra- and infratentorial embryonal tumors are moving from a traditional histology only–based approach to molecular diagnoses for risk stratification and treatment planning.7,16,22,23 In keeping with the new classification systems, there is a need to change the reference point of oncologic imaging from histopathology to molecularly integrated diagnoses.
The above numbers show that with histopathology only for diagnosis, 39% of all tumors (17 HGGs, 2 EPs, and 3 ATRTs of 56) and 71% of nonpineal tumors (16 HGGs, 2 EPs, 2 ATRTs of 28) in this study represented discrepant molecular diagnoses not intended for inclusion in this PNET trial. Our previously published results have shown markedly worse outcomes for these non-ETs7 despite a much smaller incidence of CSF dissemination by imaging and CSF cytology (5% for non-ET versus 35% for PBL/ET).
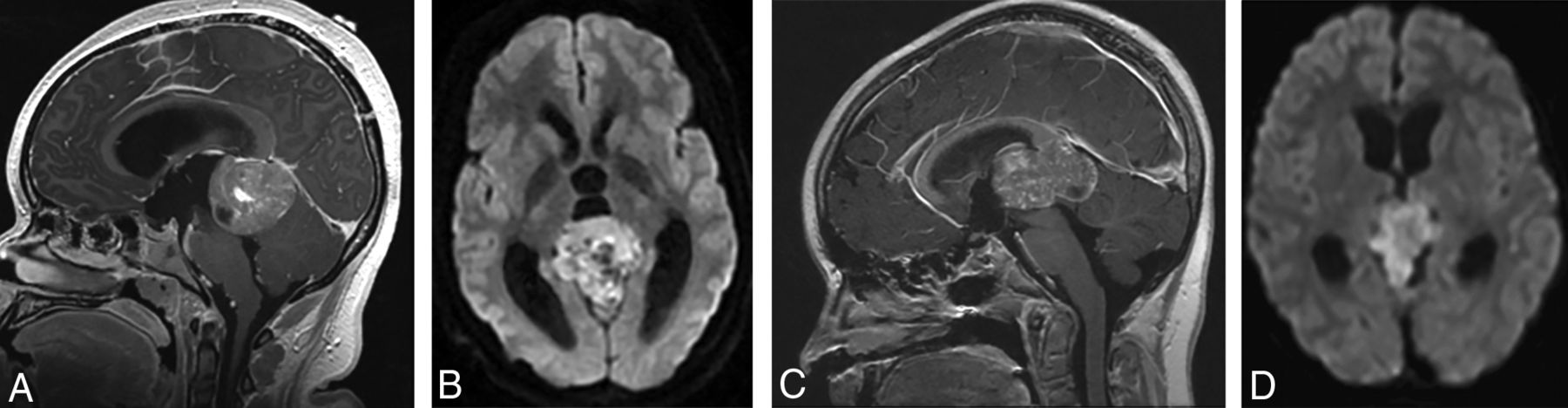
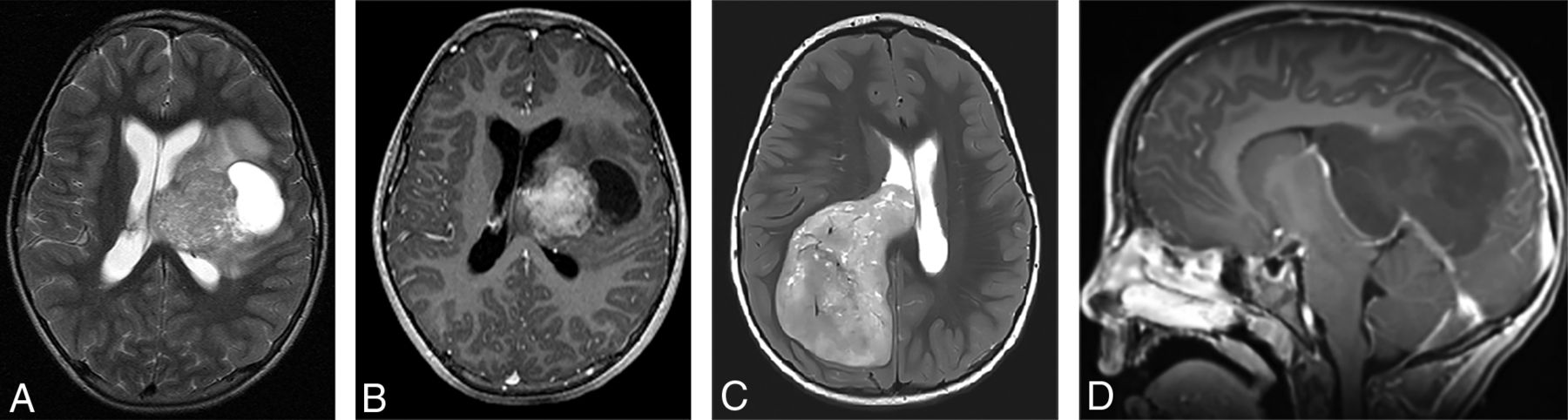
Most pineal region tumors in our study were confirmed to be pineoblastomas (Figs 3 and 4). The pineal region ATRTs in our study could not be subjectively distinguished from PBL (Fig 3). Also, there was 1 HGG in the pineal region, which, interestingly, belonged to the methylation class of diffuse midline glioma H3K27M-mutant. Of the nonpineal tumors, which include both hemispheric and ventricular locations, only 43% belonged to the embryonal group, with the remainder being HGG and EP (Table 2), with substantial overlap in the imaging appearances (Figs 5 and 6).

(A and B), A 15-year-old girl with a molecular diagnosis of pineoblastoma. Sagittal T1-weighted postcontrast (A) and axial diffusion-weighted (B) MR images demonstrate a mass centered in the pineal region with diffuse heterogeneous enhancement, small cystic foci, and diffusion restriction. (C and D), A 9-year-old girl with a molecular diagnosis of ATRT_MYC. Sagittal T1-weighted postcontrast (C) and axial diffusion-weighted (D) images demonstrate a similar mass centered in the pineal region with diffuse heterogeneous enhancement, small cystic foci, and diffusion restriction. Note the similarities in imaging appearance between the 2 examples.
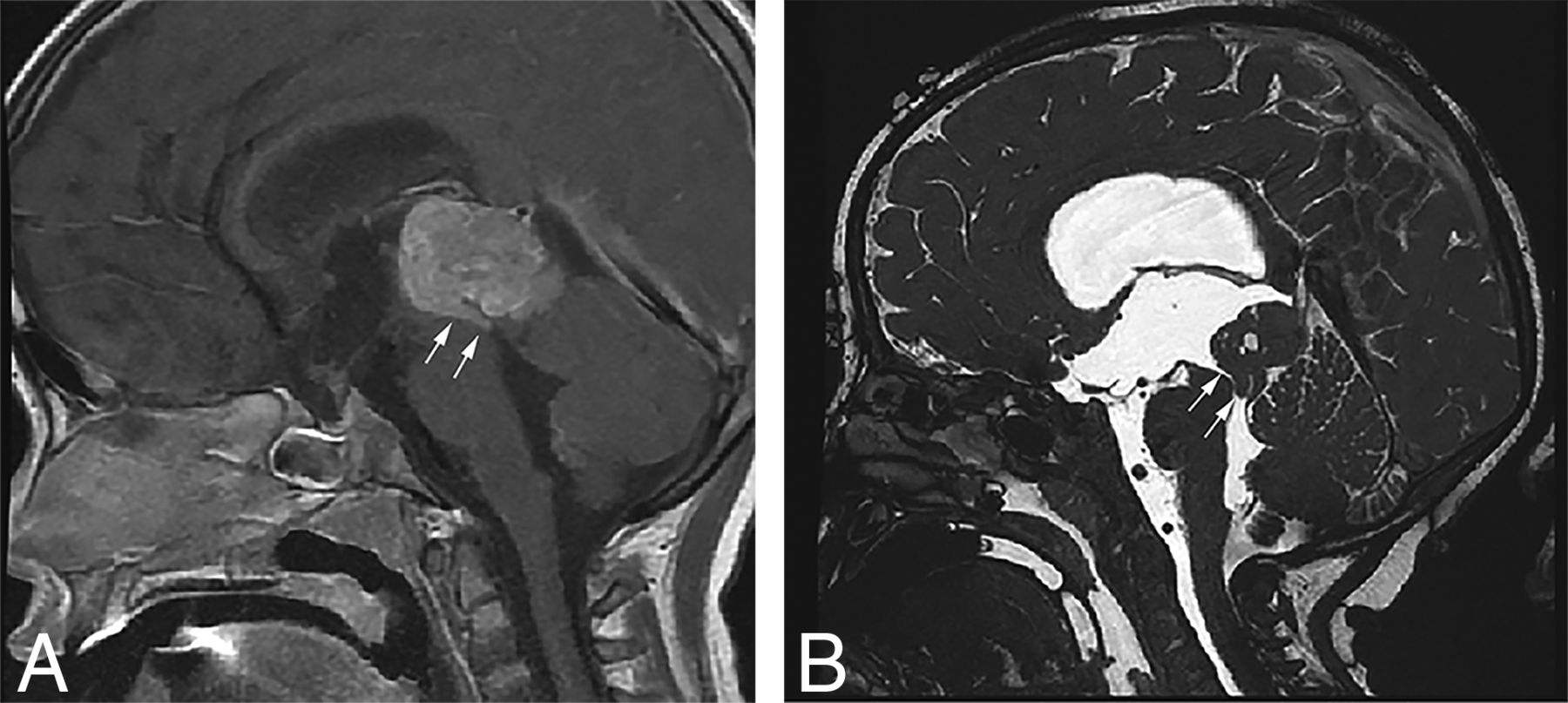
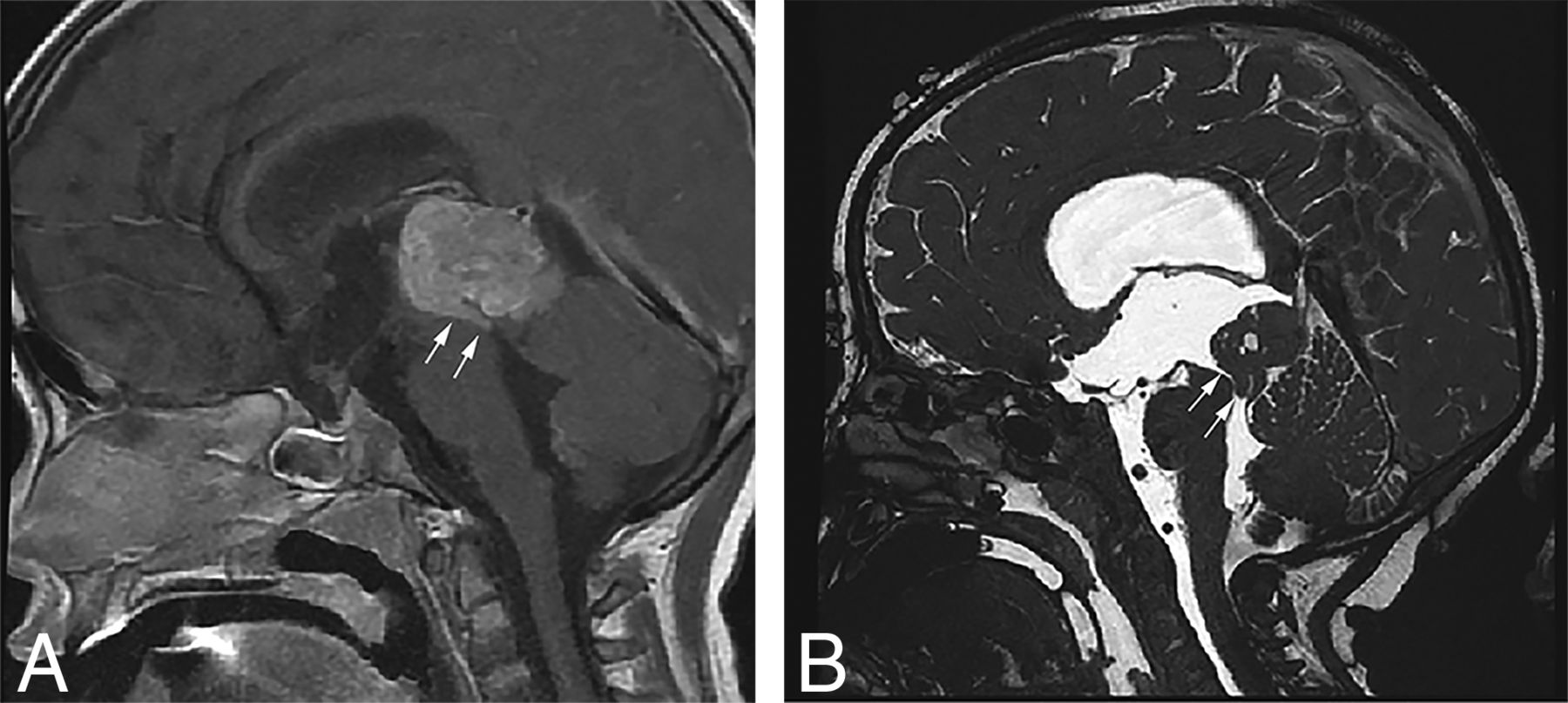
Sagittal T1-weighted postcontrast (A) and sagittal high-resolution balanced steady-state gradient-echo (B) images from 2 different patients with molecularly proved pineoblastomas demonstrating a tail-like aqueductal extension (white arrows).
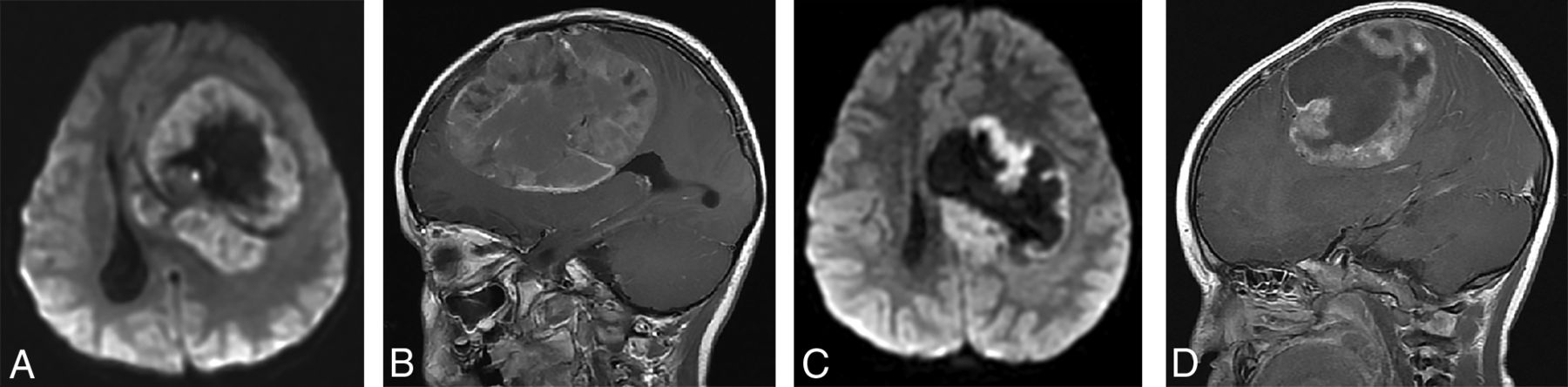
(A and B), A 4-year-old girl with a molecular diagnosis of high-grade glioma (GBM_MYCN). Axial DWI (A) and sagittal postcontrast T1-weighted (B) images demonstrate a large mass centered in the left frontal lobe with prominent necrotic/cystic areas centrally and diffusion restriction and moderate heterogeneous enhancement of the solid component. (C and D), A 4-year-old boy with a molecular diagnosis of ependymoma (EP_RELA). Axial DWI (C) and sagittal postcontrast T1-weighted (D) images demonstrate a large mass centered deep in the left hemisphere with prominent necrotic/cystic areas centrally and diffusion restriction and moderate heterogeneous enhancement of the solid component. Note the similarities in age and imaging appearance between these 2 patients with different molecular diagnoses.
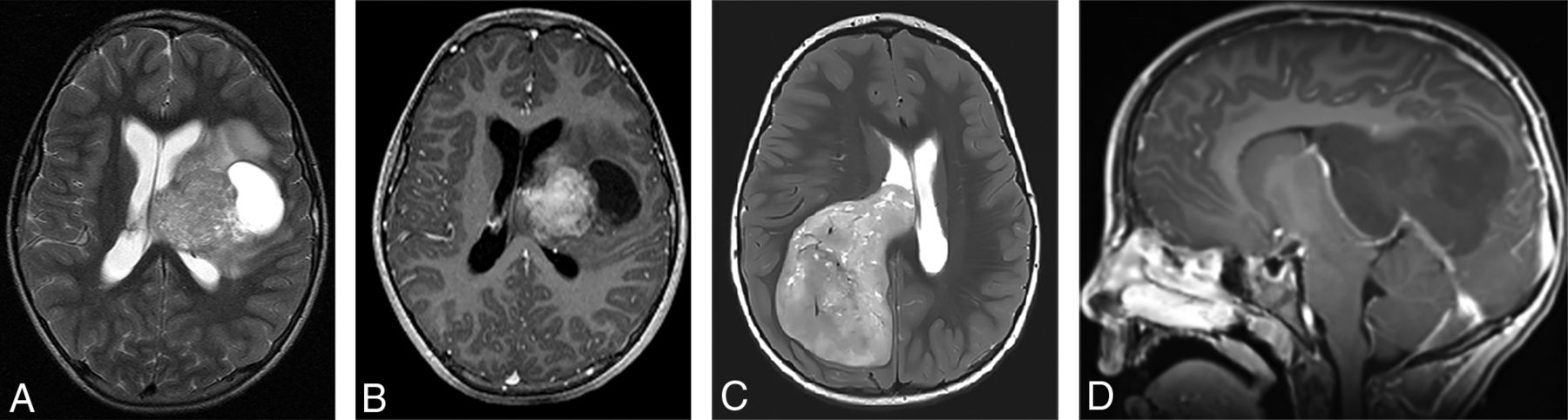
Two different patients with a molecular diagnosis of ET. (A and B), A 10-year-old girl with axial T2-weighted (A) and axial postcontrast T1-weighted (B) images has a large mass centered in the left deep nuclei with a prominent cystic component and moderate enhancement of the solid component. The tumor subclass was CNS_NB_FOXR2. Please note the similarities with high-grade glioma and ependymoma illustrated in Fig 5. (C and D), A 5-year-old girl with axial T2-weighted (C) and sagittal postcontrast T1-weighted (D) images has a large solid mass centered in the right lateral ventricle with minimal-to-no enhancement. The tumor subclass was ETMR. Of note, both of these tumors demonstrated diffusion restriction (not shown).
The median age (9 years) in this study was higher than that previously reported for supratentorial PNETs (6.5 years).4 The median age at diagnosis for pineoblastomas (8.7 years) in our study is also higher than that in a recent large series (5.5 years).24
With all locations included, the non-ETs were larger with more ill-defined margins and surrounding edema compared with the PBLs/ETs. This finding may be partly related to location, with pineal region tumors probably presenting earlier because of hydrocephalus, though tumor biology and rate of growth could be contributing factors. After we excluded pineoblastomas, none of the MR imaging parameters were statistically significant between the remaining ETs and non-ETs; however, the ETs had a tendency toward less heterogeneity and better-defined margins and occurred at a slightly younger age compared with non-ETs.
Because of the rarity of these tumors, there are only a few published reports on their imaging features.25⇓⇓–28 The PBLs in our study demonstrated MR imaging features broadly similar to those in previous descriptions, including diffusion restriction, cystic/necrotic change, and variable enhancement. Parenchymal invasion, however, was rare in our study, which conflicts with one of the prior reports.27 An “aqueductal tail” was seen in 19% of PBL (Figs 3 and 4). Other than a single case report on PBL,29 this finding has not been mentioned in any of the previous descriptions of PBL or pineal region germ cell tumors. Although such a “plastic” tumor extension is well-described for infratentorial ependymomas,30 neither of the 2 molecularly diagnosed EPs in our study were seen in the pineal region. This observation can be explored in future studies to potentially distinguish PBLs from other pineal region masses, an issue that has always been considered difficult by imaging.31,32
CNS-PNETs have typically been described on imaging as large, heterogeneous, diffusion-restricting hemispheric or ventricular masses in young children.28 No studies have compared the imaging findings with other malignant supratentorial tumors like high-grade gliomas. A report from the German multicenter HIT trial compared the MR imaging findings of ependymoblastomas and ependymomas with CNS-PNET NOS. Although the authors found some differences by imaging, their overall conclusion was that precise distinction in individual cases may not be feasible.33
The study has several limitations that must be acknowledged. Methylation profiling was not available for all enrolled patients, and some additional patients were excluded because of inadequate imaging. The MR imaging techniques were not standardized, and quantitative assessments such as ADC measurements were not performed. The patients received different treatments based on the trial protocol, and that feature could potentially confound the correlation with outcomes, though there were no significant differences in the outcome among treatment groups in our primary analysis.7 Radiologically, these supratentorial tumors can be viewed as 2 groups based on location, pineal and extrapineal; however, they were included together for analysis because of shared histopathology and their designation as a single entity by neuro-oncologists for treatment purposes. Because most of the pineal region tumors eventually were proved to be pineoblastomas and most of the nonpineal tumors were nonembryonal, including these together skewed the statistical results. We also did a separate statistical analysis of ET versus non-ET after excluding pineoblastomas, a distinction that is more of a diagnostic dilemma from a radiologic standpoint.
CONCLUSIONS
We describe the imaging features of a large cohort of histologically diagnosed supratentorial PNETs, including pineoblastomas, in correlation with the molecular diagnoses and outcomes. Knowledge of current molecularly defined tumor entities and their relative frequencies and locations will help the radiologist make more accurate predictions of the tumor types. For nonpineal tumors, a diagnosis of non-ET such as HGG or EP should be high on the list of a radiographic differential. Given the overlap of MR imaging findings, it may not be possible to offer a single diagnosis with certainty; thus, imaging does not substitute for obtaining molecular testing. However, a narrower differential diagnosis in conjunction with initial histopathology will be more helpful in guiding the surgery and radiation planning, and the criterion standard molecular testing should guide the eventual course of treatment. The current study provides a systematic description of conventional MR imaging findings in reference to the molecular diagnoses, and future studies in this direction using advanced imaging and radiomic techniques may be useful.
Footnotes
This work was supported, in part, by National Institutes of Health grants No. U10-CA180886 (National Clinical Trials Network Operations Center Grant) and U10-CA180899 (National Clinical Trials Network Statistics and Data Center) and a grant from the St. Baldrick's Foundation.
Disclosures: Alok Jaju—RELATED: Grant: Children's Oncology Group, Comments: National Institutes of Health (National Clinical Trials Network) grant U10 CA180886*; Support for Travel to Meetings for the Study or Other Purposes: Children's Oncology Group, Comments: National Institutes of Health (National Clinical Trials Network) grant U10 CA180886. David Capper—UNRELATED: Patents (Planned, Pending or Issued): patent pending for a method to classify brain tumors by DNA methylation profiling. *Money paid to the institution.
Paper previously presented, in part, at: Annual Meeting of the American Society of Neuroradiology, May 18–23, 2019; Boston, Massachusetts.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received April 8, 2019.
- Accepted after revision August 21, 2019.
- © 2019 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}