
Abstract
SUMMARY: Ectopic cerebellar tissue is a rare entity likely secondary to multiple, interacting, developmental errors during embryogenesis. Multiple sites of ectopic cerebellar tissue have been reported, including extracranial locations; however, an intracranial location is most common. We report on the MR imaging findings of a multi-institutional series of 7 ectopic cerebellar tissue cases (2 males, 4 females, 1 fetal) ranging from 22 weeks 5 days' gestational age to 18 years of age. All cases of ectopic cerebellar tissue were diagnosed incidentally, while imaging was performed for other causes. Ectopic cerebellar tissue was infratentorial in 6/7 patients and supratentorial in 1/7 patients. All infratentorial ectopic cerebellar tissue was connected with the brain stem or cerebellum. MR imaging signal intensity was identical to the cerebellar gray and white matter signal intensity on all MR imaging sequences in all cases. Ectopic cerebellar tissue should be considered in the differential diagnoses of extra-axial masses with signal characteristics similar to those of the cerebellum. Surgical biopsy or resection is rarely necessary, and in most cases, MR imaging is diagnostic.
ABBREVIATION:
- ECT
- ectopic cerebellar tissue
The term “ectopia” refers to an abnormal congenital or acquired position of an organ or tissue. Ectopic cerebellar tissue (ECT) is an extremely rare condition. Only individual case reports of ECT have been reported to date.1⇓-20
ECT can be composed of mature or immature cerebellar tissue.12 The exact mechanism of ECT development is unknown, but it is believed to be the result of an anomalous cell migration during embryogenesis.14,16 Five major mechanisms have been proposed to explain the development of ECT: 1) herniation of fetal brain tissue during embryogenesis, 2) embryonic neural rests, 3) preferential differentiation of 1 germ layer of a teratoma along neural lines, 4) true astrocytomas, and 5) similar to split spinal cord malformation pathogenesis, differentiation of pluripotent cells leading to the formation of a structure resembling the cerebellum.9,10,12,21
The goal of this article was to report a multi-institutional series of ECT cases, to discuss the common and unique imaging features of each case, and to review the existing cases in the literature. To the best of our knowledge, this is the first series of ECT cases.
Case Series
We collected cases with a diagnosis of ECT from 5 institutions for this retrospective, multicenter, international case series study. Informed consent was waived due to the retrospective nature of the study. Patient information including demographics, history, and MR imaging studies was collected. MR imaging studies were re-evaluated at Texas Children's Hospital by 2 pediatric neuroradiologists (S.F.K. and T.A.G.M.H., with 9 and 29 years of experience, respectively). Seven patients (2 males, 4 females, 1 fetal case) ranging from 22 weeks 5 days' gestational age to 18 years of age were included in this study.
Patient 1 is a 17-year-old adolescent girl with a history of gross total resection of a congenital nasopharyngeal mature teratoma after developing dyspnea after birth. In addition, a subtotal resection of a suprasellar mass lesion was performed, which was proved to be ECT without any attachment to the brain stem or cerebellum. Furthermore, an additional interhemispheric arachnoid cyst was resected when she was 4 months of age.11,12 The recovery after the operation was complicated by moderate atrophy of the right optic nerve, which was treated with temporary eye patch coverage of the healthy left eye. She underwent an additional operation for correction of her eyesight at 12 years of age. Her growth parameters were within normal limits during her follow-up visits; however, her language skills were a bit delayed, yet within the normal variation. MR imaging (Fig 1) performed at 17 years of age showed the stable, residual, suprasellar ECT.

Axial T2- (A) and T1- (B) weighted, DWI (C), ADC (D), coronal T2-weighted (E), contrast-enhanced coronal T1-weighted (F), and sagittal pre- and postcontrast enhanced T1-weighted (G and H) MR images and map of patient 1 at 17 years of age show well-defined, ectopic, cerebellar tissue (A–H, arrows) in the suprasellar region. Note that the signal intensity is similar to that of the cerebellum in all sequences, and there is no contrast enhancement on postcontrast imaging.
Patient 2 is a 25-month-old boy who was born preterm at 33 weeks via cesarean delivery secondary to maternal preeclampsia. The mother also had gestational diabetes mellitus. There were no complications during pregnancy or delivery. The patient had a benign hospital course and spent 4 weeks in the neonatal intensive care unit before being discharged. Parents first noticed problems at discharge, when they noted a profound hearing loss of the left ear. A CT scan showed an incomplete partition type I anomaly of the left inner ear. At 6 months of age, he was seen by an ophthalmologist due to concern for possible crossed eyes and was diagnosed with pseudostrabismus. He was also diagnosed with bilateral optic nerve hypoplasia, with the left side being more affected than the right side. He was then referred to an endocrinologist to rule out pituitary dysfunction, given the concern for septo-optic dysplasia, but the results of hormone and stimulation testing were normal. A brain MR imaging did not show any evidence of septo-optic dysplasia (Fig 2); however, ECT was noted superior to the vermis to the left of the midline, which was connected to the adjacent brain stem via a thin stalk. The patient has not experienced any major illnesses. Developmentally, his motor milestones are on track, and he has never required any therapy.
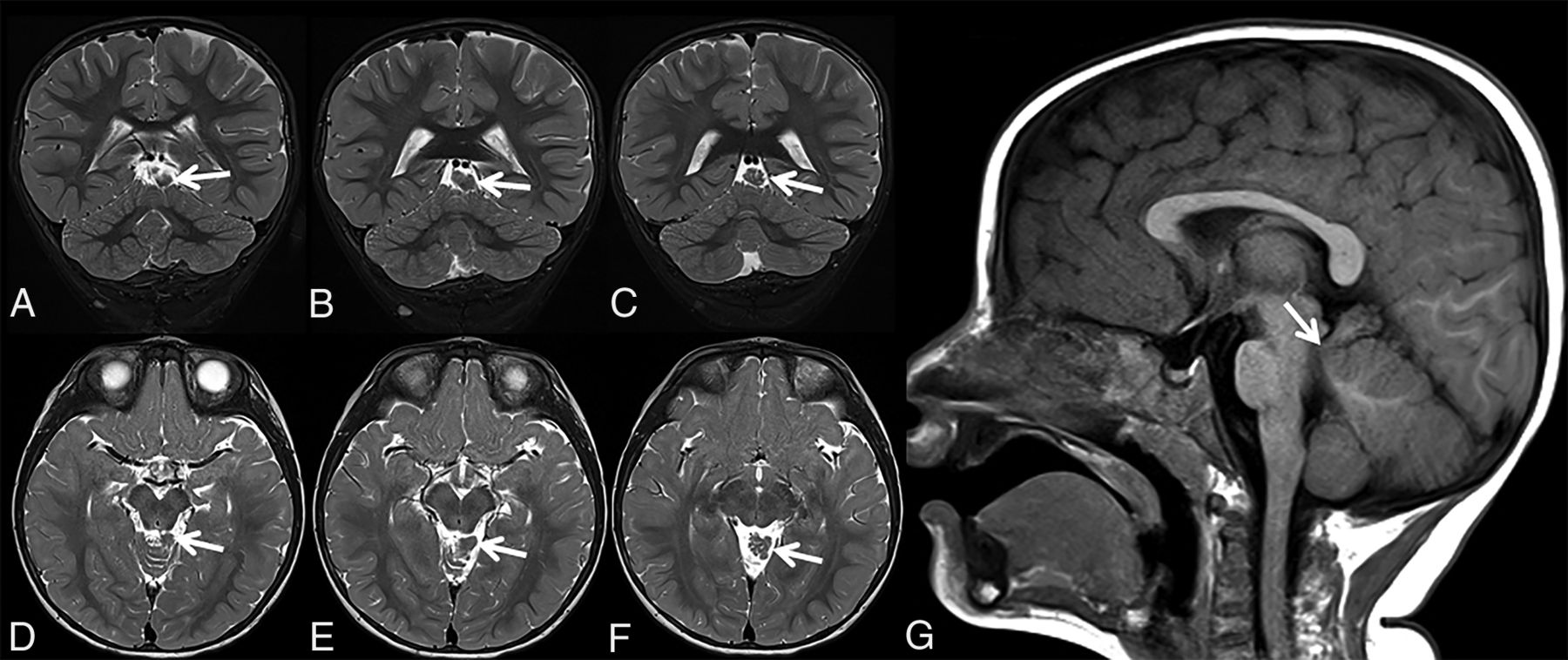
Coronal (A–C) and axial (D–F) T2-weighted and sagittal T1-weighted (G) MR images of patient 2 at 25 months of age show triangular-shaped ectopic cerebellar tissue on consecutive slices (A–F, arrows). Note the thin stalk connecting the ectopic cerebellar tissue with the brain stem (G, arrow).
Patient 3 is an 18-year-old young woman with a history of global developmental delay with intellectual disability, right sensorineural hearing loss, and simple febrile seizures. The patient developed an increasing frequency of episodes with eyes rolling back and behavioral arrest at 13 years of age. There were no preceding symptoms of the patient and mother. The episodes occurred while walking, talking, or sitting. She had no associated shaking, loss of bowel or bladder control, or facial twitching. Further work-up with MR imaging, electroencephalography, and metabolic lab tests was performed to rule out the possibility of seizures and to further investigate the etiology for her intellectual disability/developmental delay. The findings of electroencephalography were abnormal, consistent with a diagnosis of a primary generalized epilepsy. Metabolic lab test results were within normal limits. MR imaging at 13 years of age showed ECT as a well-defined right cerebellopontine angle lesion, which was stable on annual follow-up MR imaging (Fig 3).

Axial T1- (A and F) and T2- (B and G) weighted, axial DWI (C and H), ADC (D and I), and coronal T2- (E) and T1- (J) weighted MR images and maps of patient 3 at 13 years of age (upper row, arrows) and 18 years of age (lower row, arrows). The patient had a history of global developmental delay with intellectual disability, right sensorineural hearing loss, and simple febrile seizures. MR imaging shows well-defined ectopic cerebellar tissue in the right cerebellopontine angle (A–D, arrows), which was stable on follow-up MR imaging (E–H, arrows). Note the thin stalk connecting the ectopic cerebellar tissue with the middle cerebellar peduncle (E and J, arrows).
Patient 4 is a 32-month-old girl who was born at term in good condition and with all growth parameters between the 25th and 50th percentiles. She showed a delay in reaching her motor and language skills. At 32 months of age, she presented with microcephaly (45.5 cm, below the second percentile), minor facial dysmorphic features, and mild postural ataxia. MR imaging revealed a ECT along the superior/lateral contour of the right cerebellar hemisphere (Fig 4). During follow-up, the patient developed a complex neuromotor disease with dyskinetic movement disorder, ataxia, pyramidal signs in the lower limbs, a language disorder, and mild cognitive impairment; mild asymmetry was observed with the left side being more involved than the right. Findings of the follow-up MR imaging scans were stable when the girl was 4 and 6 (Fig 5) years of age. Other specific examinations performed included an electroencephalogram, which revealed, at 6 years of age, spike temporal waves while falling asleep and spike waves in the fronto-centro-temporal areas and vertex during sleep.

Coronal (A–D) and sagittal T2-weighted (E–G) MR imaging of patient 4 at 32 months of age shows a distorted/dysmorphic superior portion of the right cerebellar hemisphere, which also shows folial and sulcal anomalies. The dysmorphic superior portion also displaces the adjacent vermis. Ectopic cerebellar tissue (A–G, arrows) is connected to the cerebellum. Its MR signal is isointense with the cerebellum, and its morphology is elongated with irregular borders. Additional brain malformations included temporal lobe dysgyria and malrotated hippocampi (not shown).

Follow-up coronal and sagittal T1-weighted MR imaging of patient 4 at 6 years of age shows a stable imaging pattern of the ECT (arrows) with respect to the already-described findings in Fig 4.
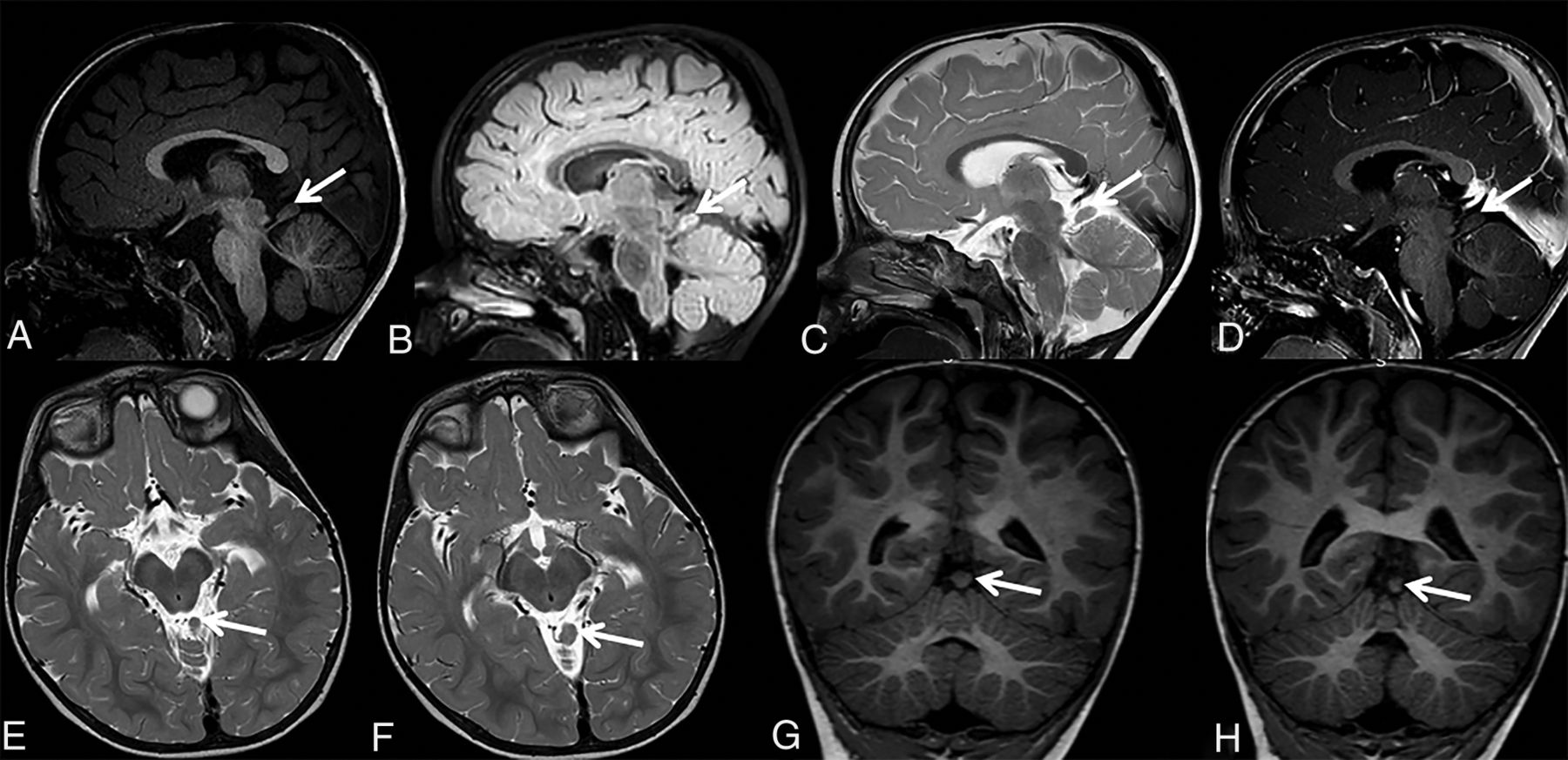
Patient 5 is a 4-year-old girl who was referred for postnatal assessment of antenatally detected ventriculomegaly, which appeared stable on postnatal follow-up sonography. She was developing normally, reaching all developmental milestones. Her head circumference was following a normal growth curve, and her extraocular movements were normal. MR imaging acquired at 3 years of age showed a thin stalk arising from the midbrain, which extended to a triangular-shaped ECT superior to the right anterior cerebellar hemisphere, with an arachnoid cyst posteriorly (Fig 6). The findings were unchanged on follow-up MR imaging 1 year later.

Sagittal T1- (upper row) and T2-weighted (lower row) MR images of patient 5 at 3 years of age show a thin stalk arising from the pontomesencephalic junction below the right inferior colliculus dorsolaterally, which extends to triangular-shaped ectopic cerebellar tissue (A–F, arrows) superior to the right anterior cerebellar lobe, with an arachnoid cyst (A–F, asterisks) posteriorly. The sagittal oblique reformat T1-weighted MR imaging shows the continuous stalk (G, arrows) between the triangular-shaped ECT and the brain stem. The findings were unchanged 1 year later (not shown).
Patient 6 is a 5-year-old boy who was born preterm at 34 weeks with moderate respiratory problems necessitating ventilatory support during the first few days of life. Otherwise, he had an uncomplicated early neonatal period and uncomplicated feeding; he was thriving with no signs of infection. Neurologic examination showed a slanting posture of his head (turning left/slanting right), which was more prominent from 1.5 to approximately 3 years of age. Since then, neurologic examination findings were regressive in frequency and intensity; association with strabismus was suspected, presently hardly seen, as well as congenital alternate strabismus divergens and a slight ptosis of the left eye. The patient showed delayed motor, language, and cognitive development. MR imaging performed at 17 months of age revealed ECT connected to the brain stem by a thin stalk (Fig 7).

Sagittal T1-weighted (A), sagittal FLAIR (B), sagittal T2-weighted (C), sagittal contrast-enhanced T1-weighted (D), axial T2-weighted (E and F), and coronal T1-weighted (G and H) MR images of patient 6 at 17 months of age show ECT connected with the brain stem by a thin stalk (A–H, arrows). Most interesting, the MR imaging appearance of ectopic cerebellar tissue in this patient is almost identical to the MR imaging findings of patient 2.
Patient 7 was a fetus of 22 weeks 5 days' gestational age after termination of pregnancy due to prenatal diagnosis of vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities (VACTERL) association. Macroscopic examination of the brain and postmortem 3T fetal MR imaging showed an incidental finding of a small appendage with a ribbed surface located superior to the left cerebellar hemisphere and originating from the left dorsal pontomesencephalic junction (Fig 8). Histologic analysis revealed this structure to be of cerebellar origin with distinguishable external granular and molecular layers and developing Purkinje cells and an internal granular layer.

Postmortem sagittal (A) and axial (B) T2-weighted fetal MR images at the gestational age of 22 weeks 5 days and a matching macroscopic postmortem image (C) of the brain show an incidental finding (A and B, arrows) of a small appendage with a ribbed surface located superior to the left cerebellar hemisphere, originating from the left dorsal pontomesencephalic junction. Histologic analysis (D) reveals that this structure is of cerebellar origin with distinguishable external granular and molecular layers and developing Purkinje cells and an internal granular layer. Adapted with permission.23
All cases of ECT were diagnosed incidentally when imaging was performed for other causes or by symptoms that were directly attributable to brain stem or cerebellar dysfunction. The Online Supplemental Data summarize the neuroimaging findings of the patients. ECT was infratentorial in 6/7 patients and supratentorial in 1/7 patients. All infratentorial ECTs were connected with the brain stem or cerebellum. ECT volume compared with the cerebellar volume was 20%–30% in 1/7 patients, 10%–20% in 2/7 patients, and 1%–10% in 4/7 patients. MR imaging signal intensity was identical to that of the cerebellar gray and white matter on all MR images. In addition, none of the patients had a concomitant brain stem or spinal/craniocervical junction malformation.
DISCUSSION
We report 7 cases of ECT. All cases of ECT (7/7) were diagnosed incidentally, when imaging was performed for other causes. Most cases were located infratentorially (6/7) and had a normal cerebellar morphology (6/7). In 1 patient (patient 1), the ECT was located supratentorially, and 1 patient (patient 4) showed an abnormal cerebellar morphology (Online Supplemental Data). Similar to our results, all previous cases (20/20) were diagnosed incidentally when imaging was performed for other causes, and most reported cases (10/15) in the literature had a normal cerebellar morphology (Online Supplemental Data).
ECT was located intracranially in all our cases. However, we found previous case reports identifying ECT in the ovaries, orbits, spinal cord, or occipital bone (Online Supplemental Data).2,4,5,7⇓⇓-10,17,20 On the basis of the wide spectrum of clinical, imaging, and histopathologic findings in the previously reported ECT cases, multiple mechanisms are likely involved in the development of ECT.12,13
The exact mechanism of ECT development remains currently unknown. However, 5 major mechanisms proposed in previous reports include the following: 1) herniation of fetal brain tissue during embryogenesis, 2) embryonic neural rests, 3) preferential differentiation of 1 germ layer of a teratoma along neural lines, 4) true astrocytomas, and 5) similar to split spinal cord malformation pathogenesis, differentiation of pluripotent cells leading to the formation of a structure resembling the cerebellum.9,10,12,21 From a histopathologic point of view, true ECTs should be distinguished from the more frequent focal cerebellar cortical dysplasias.12 Focal cerebellar cortical dysplasias are more prevalent and usually small and, in most cases, consist of a single cell type or poorly organized mixed-cell clusters.12 True ECTs are usually larger lesions composed of organized mature or maturing cerebellar tissue.12
The isthmic organizer is located below the mesencephalon at the pontomesencephalic junction and provides structural polarity of adjacent regions and organizes mesencephalic and cerebellar development. The isthmic organizer signaling is essential for cerebellar development.22 We observed 3 different types of ECTs in our case series: 1) suprasellar ECT (patient 1), 2) ECT sitting on a small stalk connected with the adjacent brain stem (patients 2, 5, 6, and 7), and 3) ECT sitting on a cerebellar hemisphere (patients 3 and 4). In the second group (patients 2, 5, 6, and 7), the stalk and ECT were all located in the region of the isthmic organizer. An alteration of isthmic organizer signaling during embryogenesis may possibly explain the pathogenesis of these cases.
Two of our patients had associated arachnoid cysts (Online Supplemental Data). Patient 1 had an interhemispheric arachnoid cyst, which was removed surgically during the neonatal period, and patient 5 had an arachnoid cyst adjacent to the ECT. The association of arachnoid cysts and a CSF collection adjacent to ECT may indicate the importance of meningeal stimulus/interaction during cerebellar development.16 During formation of the normal cerebellum, the germinal tissue possibly migrates through the ependymal cell lines into the CSF. Therefore, obstruction of normal flow of the CSF may result in ECT, with inflammation of the arachnoid membrane and an arachnoid cyst.
Differentiating ECT from low-grade tumors by neuroimaging is necessary to avoid unnecessary surgery, especially in cases centered at the vulnerable cerebellopontine angle (patient 3). Distinguishing MR imaging features include a lack of contrast enhancement of the lesion, signal characteristics similar to those of the adjacent cerebellum on all MR imaging sequences, diffusion characteristics similar to those of the normal cerebellum, a sulcation pattern mimicking the normal cerebellum, a thin parenchymal stalk that connects the ECT to the adjacent cerebellum or brain stem, and a stable appearance on follow-up imaging without any sign of abnormal, noncommensurate growth.18
In conclusion, ECT is a rare entity, which is attributed to possible multiple, interacting, developmental errors during embryogenesis. Multiple locations were reported; however, the intracranial and infratentorial locations appear most common. ECT should be considered in the differential diagnoses of extra-axial masses with signal characteristics similar to those of the cerebellum. Surgical biopsy or resection is rarely necessary; in most cases, MR imaging is diagnostic.
Footnotes
Disclosures: Roberta Battini—UNRELATED: Board Membership: Teva Pharmaceutical Industries for trial ongoing on dyskinetic cerebral palsy, Ionis Pharmaceuticals for new genetic treatment on Angelman syndrome.
References
- Received November 10, 2020.
- Accepted after revision January 5, 2021.
- © 2021 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.