
SUMMARY:
Monoclonal antibodies have become increasingly popular as novel therapeutics against a variety of diseases due to their specificity, affinity, and serum stability. Due to the nearly infinite repertoire of monoclonal antibodies, their therapeutic use is rapidly expanding, revolutionizing disease course and management, and what is now considered experimental therapy may soon become approved practice. Therefore, it is important for radiologists, neuroradiologists, and neurologists to be aware of these drugs and their possible different imaging-related manifestations, including expected and adverse effects of these novel drugs. Herein, we review the most commonly used monoclonal antibody–targeted therapeutic agents, their mechanism of action, clinical applications, and major adverse events with a focus on neurologic and neurographic effects and discuss differential considerations, to assist in the diagnosis of these conditions.
ABBREVIATIONS:
- Ab
- antibody
- AE
- adverse event
- Ag
- antigen
- ARIA
- amyloid-related imaging abnormalities
- CAA
- cerebral amyloid angiopathy
- CTLA-4
- cytotoxic T-lymphocyte Ag 4
- Fc
- constant fragment
- IRAE
- immune-related AE
- IRIS
- immune reconstitution inflammatory syndrome
- mAb
- monoclonal antibody
- NTZ
- natalizumab
- PML
- progressive multifocal leukoencephalopathy
- PRES
- posterior reversible encephalopathy syndrome
- TNF
- tumor necrosis factor
Antibodies (Abs) are produced by B cells and share the same basic structure, which is Y-shaped and allows Ab molecules to carry out their dual functions: antigen (Ag) binding and biologic activity mediation.
The base of the Ab is composed of a constant fragment (Fc), which imparts the effector properties of the molecule in the immune system, on its recognition by Fc receptors present on various cell types that determine the actual biologic effect. The opposing end is the variable fragment of the molecule that binds to unique Ags, imparting the specificity of each Ab. Each B-cell line produces Abs specific for 1 unique epitope, thus resulting in monoclonal Ab (mAb) production. At times, multiple B cells generate different Abs against multiple epitopes from different regions of a full-length protein Ag. This feature allows immune responses to a highly specific target to disable/eliminate the Ag from the system, after which B cells remain in the bloodstream ready to produce Abs when the Ag is encountered again.1
The method of producing various mAbs against a variety of targets, ranging from cancer to autoimmune diseases, is similar. The target Ag, or part thereof, is injected into a host animal, mounting an immune response and creating a pool of B cells specific to that Ag. The B cells are then harvested and cultured with myeloma cells, creating a hybridoma of a line of immortal mAb-producing cells that can divide to generate more of the desired mAbs. However, the Fcs of Abs produced in host animals are foreign to the human immune system, inducing a response that can neutralize the treatment or even lead to an anaphylactic reaction. Three options are available to overcome this hurdle: The mAbs can be humanized through the use of transgenic mice that create Abs with human Fcs. Products that use this method have names that end in -zumab.1 A second approach is to split host-derived mAbs and then combine the created variable fragment with human Fc. The end products are chimeras and have names that end in -ximab.1 A third approach is to create a fusion protein composed of a human Fc and an Ag-specific receptor. These do not resemble typical Abs but act in a similar manner and have names that end in -cept.1 mAbs mediate their therapeutic effect by targeting cells for death via Ab-induced apoptosis, Ab-dependent cell cytotoxicity, or complement mediated cell lysis, or mAbs can physically block a receptor ligand interaction,1 with the ultimate goal of targeting a specific cell population or molecule relevant to disease pathogenesis.
Generally, only ∼0.1% of the circulating Abs enter the brain, and they do so via either adsorptive-mediated endocytosis, carrier-mediated transport, or receptor-mediated transcytosis.2 This process is discussed in greater detail in the Online Supplemental Data.
Immunostimulating mAbs: Ipilimumab and Tremelimumab
Ipilimumab and tremelimumab are mAbs that selectively block cytotoxic T-lymphocyte Ag 4 (CTLA-4), an immune-inhibitory protein expressed on activated T cells, thereby enhancing the immune response against tumors.3 In multiple trials, they have shown high efficacy in the treatment of metastatic melanoma.3,4 They are currently under study for the treatment of various other types of advanced malignancy, including metastatic renal cell carcinoma and prostate cancer.
The most common adverse events (AEs), affecting >10% of patients, are diarrhea, rash, pruritus, fatigue, nausea or vomiting, abdominal pain, insomnia, anorexia, hallucinations, and temperature intolerance.3,4 In addition, a novel spectrum of autoimmune-inflammatory toxicities surfaced, the pathogenic mechanism of which seems to be sustained by the positive modulation on the immune system known as immune-related AEs (IRAEs).5 The gastrointestinal tract, liver, skin, pituitary, and the musckuloskeletal system are most frequently involved, leading to colitis, hepatitis, dermatitis, hypophysitis, and arthritis. Rarer IRAEs include uveitis, thyroiditis, primary adrenal insufficiency, polyneuritis, Guillain-Barré syndrome, optic ischemic or peripheral neuropathy, pneumonitis, pancreatitis, aseptic meningitis, nephritis, red blood cell aplasia, myocarditis, myasthenia gravis, sarcoidosis, and myositis. The frequency and severity of IRAEs appear to be dose-dependent.6,7 Occasionally (∼1%), deaths have occurred as a result of colonic perforation.8
From a neurologic and neuroradiologic point of view, hypophysitis is the most commonly encountered AE of these drugs and is estimated to occur in 10%–18% of patients treated.9 MR imaging features of hypophysitis are typically characterized by diffuse enlargement of the pituitary gland, with loss of normal posterior pituitary signal intensity on the precontrast T1WI and variable enlargement of the infundibulum. Enhancement is typically uniform but can be heterogeneous (Fig 1).10
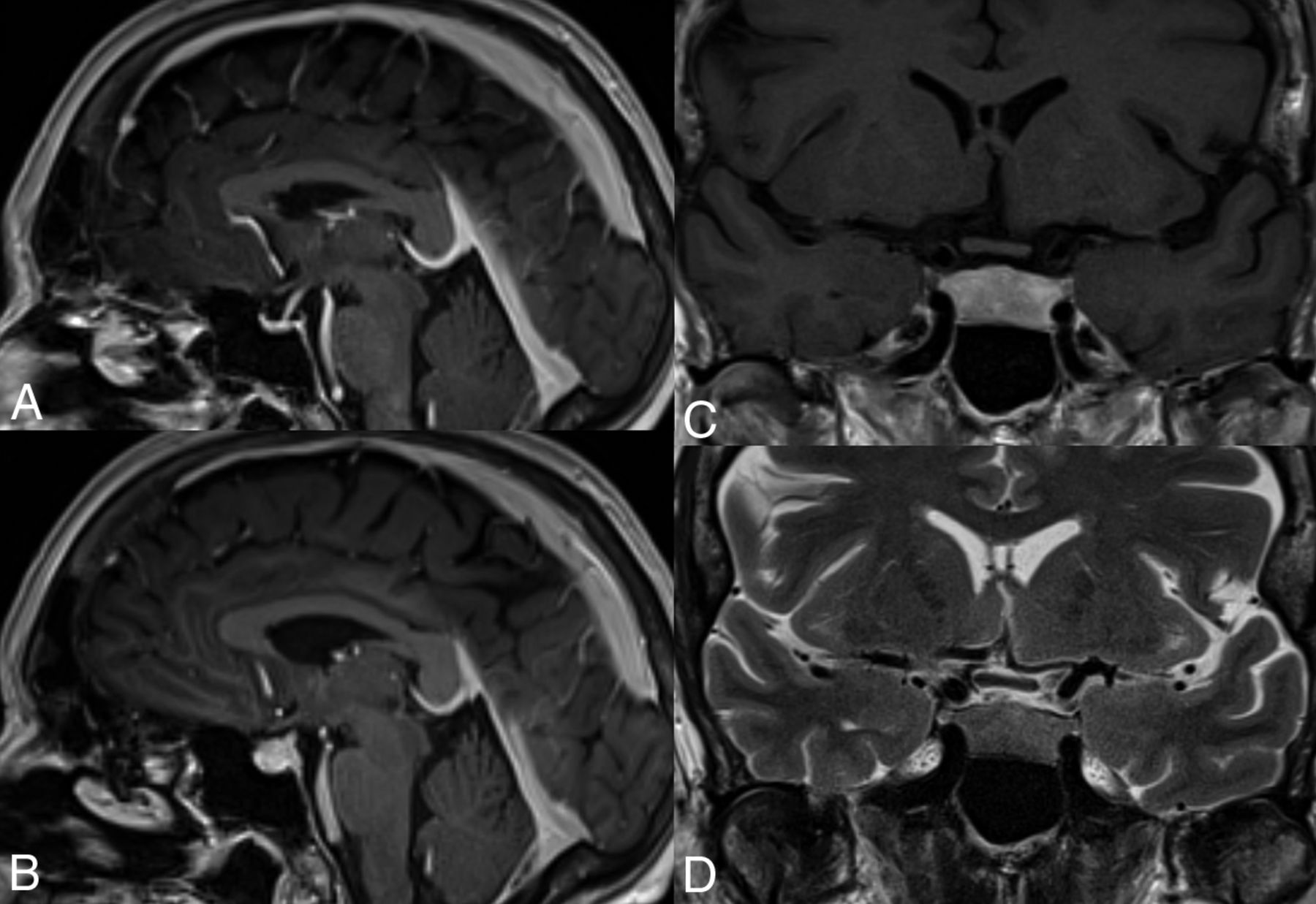
Hypophysitis induced by anti-CTLA-4 mAbs. This 61-year-old woman with metastatic melanoma underwent adjuvant treatment with ipilimumab. A, Sagittal T1 fast-spoiled gradient recalled image cut through the midline, postgadolinium, at initiation of treatment in the patient with a normal-for-age pituitary gland. MR imaging 6 months later (B–D) demonstrates a markedly thickened, densely enhancing pituitary gland in keeping with ipilimumab-induced hypophysitis.
Symptoms are variable, such as fatigue, insomnia, anorexia, hallucinations, and temperature intolerance. The laboratory findings may show a decline in values of biomarkers of the affected pituitary lobe, ie, thyroid-stimulating hormone, cortisol, and sex hormones compared with the normal ranges.
Several issues concerning anti-CTLA-4–induced hypophysitis remain to be fully elucidated, including the higher prevalence in males and the lower incidence in patients exposed to tremelimumab compared with ipilimumab.11
Clinically, resolution of acute symptoms after discontinuation of mAb treatment and steroid therapy is typically seen; if required, hormone replacement therapies are initiated. It remains to be seen how many patients will have persistent partial hypopituitarism or panhypopituitarism requiring long-term hormone replacement. Most important, the treatment of IRAEs with immunosuppressive agents, such as corticosteroids, does not appear to affect the antitumor response.6
The most important differential diagnosis is that of a new pituitary metastatic deposit; this can only be evaluated with time with follow-up imaging after cessation of mAb treatment (Online Supplemental Data). Other causes of hypophysitis unrelated to anti-CTLA-4 mAbs may be considered, including polyglandular autoimmune syndromes and immunoglobulin G–related systemic disease, secondary hypophysitis due to local inflammation of the pituitary as a reaction to sellar disease, or systemic diseases (infectious or inflammatory disorders, eg, Wegener granulomatosis, sarcoidosis, tuberculosis, or syphilis).12,13
Immunosuppresive mAbs: Natalizumab
Natalizumab (NTZ) is a humanized monoclonal antibody directed against the α4β1 and α4β7 integrins. It prevents inflammatory cells from binding to cerebrovascular endothelial cells, thereby preventing them from crossing the BBB,14 resulting in CNS immunosuppression. It is mainly used for relapsing-remitting MS but has also proved efficacy in Crohn disease and ankylosing spondylitis.15
The overall incidence of AEs associated with NTZ is low. However, 3 associated phenomena need to be kept in mind because early recognition of their spectrum of clinical and imaging findings is crucial to limit morbidity: The primary AE is progressive multifocal leukoencephalopathy (PML). Secondarily, on clearance of NTZ, a PML-associated immune reconstitution inflammatory syndrome (PML-IRIS) may occur, resulting in a paradoxical worsening of symptoms. Third, on cessation of NTZ, an exuberant rebound of MS may be observed.16
PML
The cause of PML is the neurotrophic polyoma JC virus. By causing lytic infection of brain oligodendrocytes and, to a lesser extent, astrocytes, widespread CNS demyelination ensues. Clinical signs and symptoms of PML vary. The most common symptoms in PML are confusion, hemiparesis, incoordination, speech disturbance, and visual problems.17
On MR imaging, PML presents with preferentially peripheral, variably sized and shaped white matter T2/FLAIR hyperintensities, classically involving subcortical U-fibers, which do not conform to cerebrovascular territories and do not enhance (Fig 2). They are bilateral, with an asymmetric distribution, growing larger and becoming confluent as the disease progresses, with no or only mild mass effect. Involvement of the overlying cortex, while originally thought to be rare, has been increasingly reported.18,19

NTZ-induced PML. PML in a 52-year-old male patient with Crohn disease treated with NTZ. FLAIR (A and F), enhanced T1 (B and G), SWI (C and H), DWI (D and I), and ADC maps (E and J) at ganglionic (A–E) and supraganglionic (F–J) levels demonstrate the typical imaging features of PML with subcortical U-fiber involvement, lack of enhancement, and asymmetric involvement of the white matter.
With time, lesions become increasingly hypointense on T1WI as irreversible white matter destruction occurs, and they may demonstrate a “microcyst” or “granular” pattern,20 which may represent small areas of demyelination in the immediate vicinity of infected oligodendrocytes.18 These will lead with time to progressive brain atrophy (Online Supplemental Data).
Superficial and deep gray matter involvement (more commonly the thalami) has been reported in conjunction with white matter lesions in up to 5%–31% of patients.21 Posterior fossa involvement is most commonly in the cerebellum and middle cerebellar peduncles, but the brainstem can also be involved with crescent-shaped lesions.22 The optic nerve and spinal cord are spared, and hemorrhage is a rare finding of PML but has been reported in patients with HIV taking NTZ.23,24
Enhancement in NTZ-associated PML may indicate a worse prognosis and may be patchy, linear, nodular, or peripheral.25
The DWI and DTI appearance of PML lesions varies depending on the stage of the disease. Early on, when lesions are relatively small, infected oligodendrocytes swell and die, resulting in high signal on DWI.16 Fractional anisotropy values are reduced, compatible with myelin injury. As the lesions enlarge, the signal on DWI remains high within the periphery as new oligodendrocytes become infected.26 When lesions become quiescent, the rim loses its DWI hyperintensity. In later phases of tissue destruction, a relatively free diffusion of water within the irreversibly damaged tissue is observed.9
A confirmed diagnosis of PML is based on histologic examination or CSF polymerase chain reaction.
Differential diagnoses of PML include other opportunistic viral infections, including varicella zoster encephalomyelitis and herpes simplex encephalitis and various bacterial and fungal infections. Varicella zoster myelitis may demonstrate T2-hyperintense signal and enhancement in the spinal cord,27 differentiating it from PML. Herpes zoster encephalitis demonstrates rapidly progressive cortical and subcortical T2 hyperintensity, swelling, and occasional enhancement involving the limbic regions.28
MS relapse is presumably the most important differential for new lesions. New MS lesions tend to be small, focal, and well-delineated, favoring the periventricular and juxtacortical white matter and are typically round or ovoid.20,21 MS lesions may enhance homogeneously or peripherally, whereas PML generally does not. MS lesions generally restrict diffusion only in the hyperacute phase (<1 week).29 Both tumefactive demyelinating and PML lesions demonstrate large areas of T2 hyperintensity and T1 hypointensity; however, mass effect is usually greater with the former, and T1 hypointensity improves with time in tumefactive lesions due to remyelination,30 while improvement does not occur in PML.
Acute disseminated encephalomyelitis can appear similar to PML, with large areas of T2 signal abnormality in the white matter and deep gray structures, with minimal enhancement and variable mass effect.31
Posterior reversible encephalopathy syndrome (PRES) can appear similar to PML on initial examination, but lesions tend to be more symmetric than those in PML and predominantly involve the posterior aspects of the brain, and lesions typically resolve with treatment of the inciting etiology. In addition, PRES-associated T2/FLAIR hyperintensities classically present with facilitated diffusion.
The goal of treatment for NTZ-associated PML is the restoration of immune function by rapid removal of the drug, typically achieved with plasma exchange or immunoadsorption, to clear the drug from the α4β1 receptors.32
PML-IRIS
Once NTZ is cleared by plasma exchange, many (∼70%)33 patients with prior PML will experience rapid progression of neurologic symptoms, thought to be due to an exuberant immune response to viral Ags resulting in inflammation-mediated damage to infected and noninfected neuronal and glial tissue.
PML-IRIS may also occur following discontinuation of NTZ, usually ∼90 days after the last dose, reflecting the longer time necessary to clear the drug without plasma exchange.16
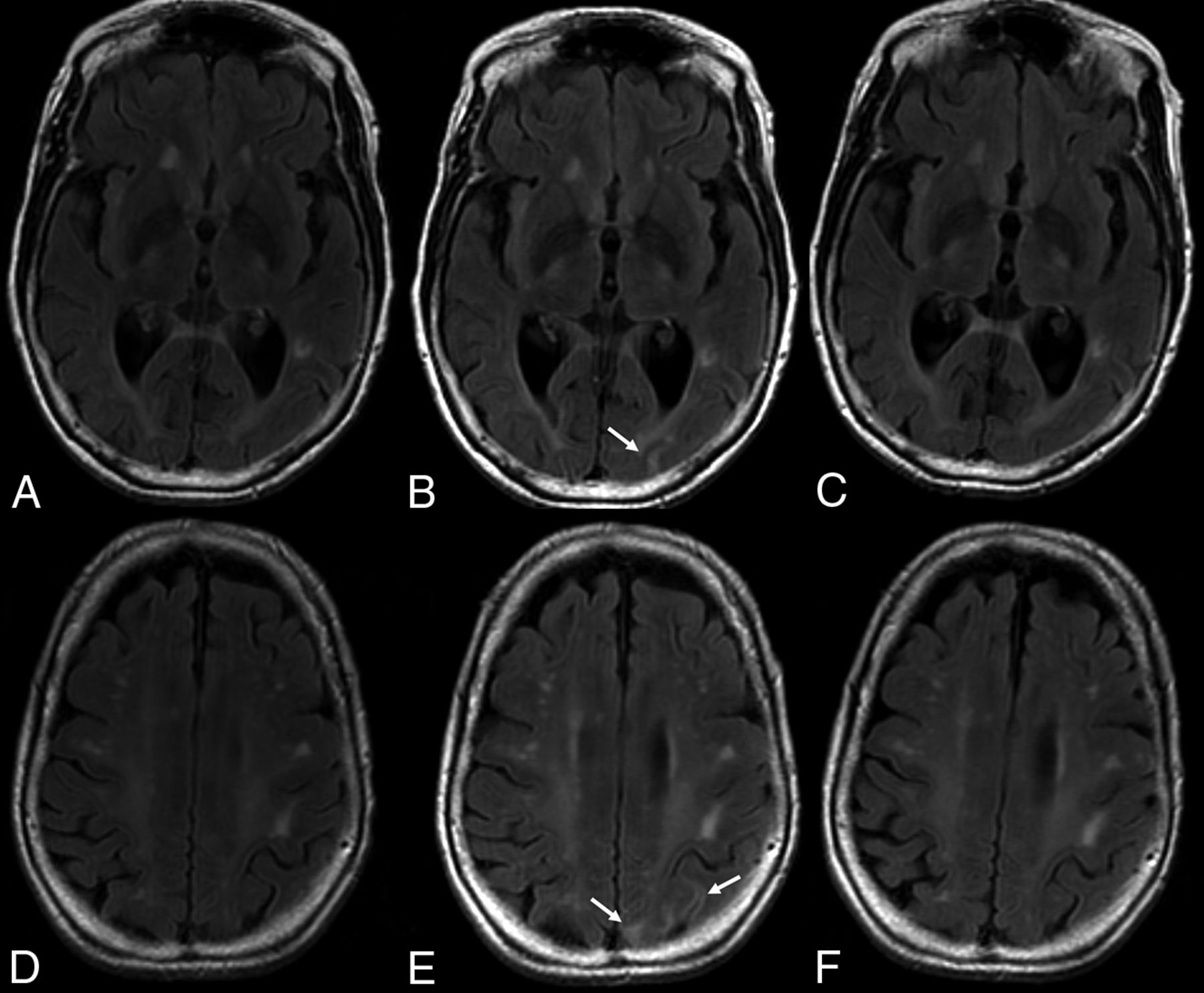
On imaging, pseudoprogression of the PML imaging abnormalities with active inflammation and new peripheral and open rim enhancement is seen (Fig 3). Existing PML lesions may increase in size or coalesce as more white matter becomes involved, accompanied by increasing cerebral swelling and mass effect. Contrast enhancement develops or increases (variable patterns: patchy, punctate, irregular, and hazy, ill-defined).18 Across time, T1 hypointensity increases, indicating irreversible white matter damage with long-term atrophy of the overlying cortex due to retrograde neuronal degeneration. The therapy for PML-IRIS is high-dose corticosteroids.34
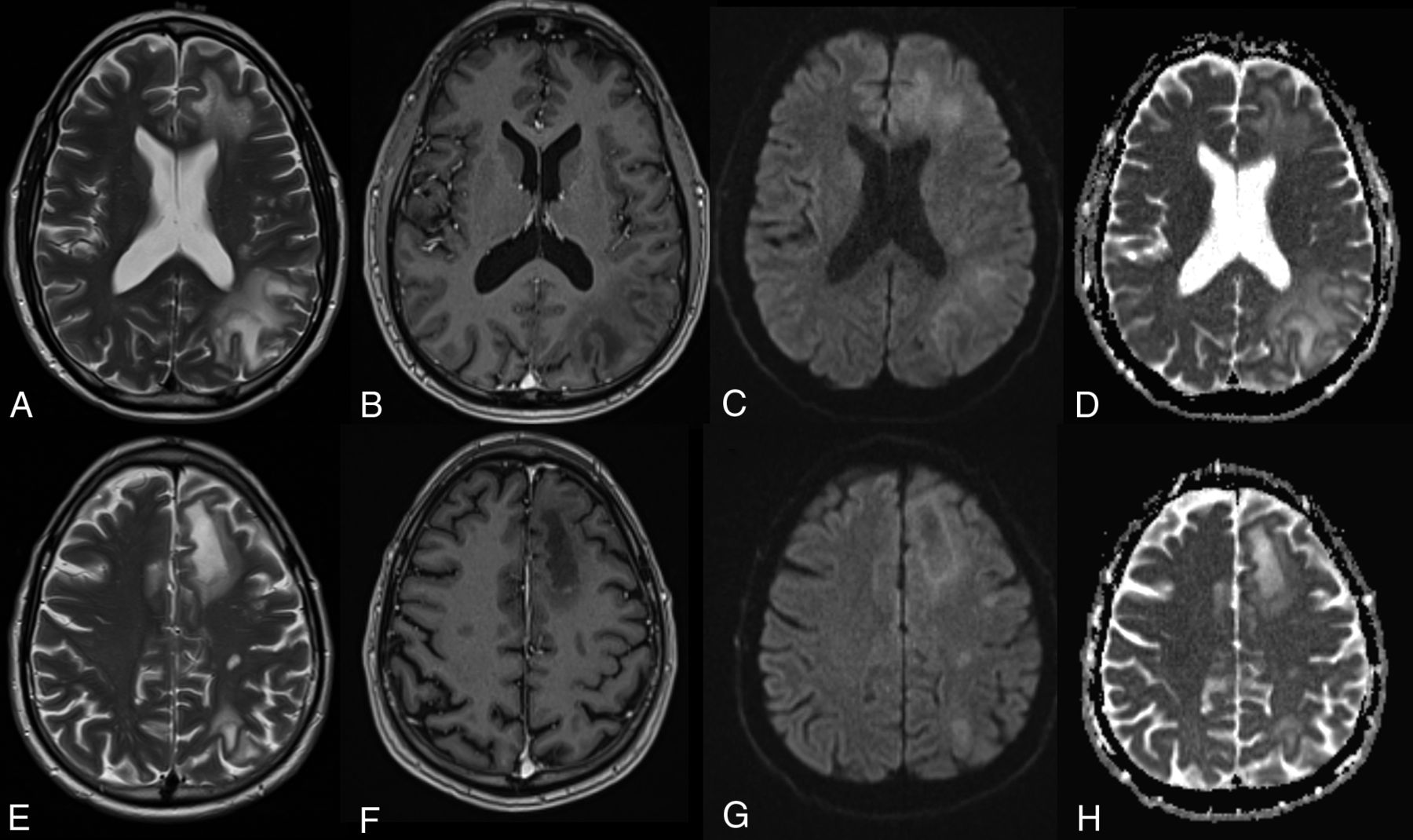
PML-IRIS after cessation of NTZ treatment. PML-IRIS in the same patient as depicted in Fig 2 following cessation of NTZ. The condition of the patient deteriorated clinically, prompting additional imaging that now demonstrates a “leading edge” of demyelination toward the white matter, mild enhancement, and DWI hypersignal, in keeping with cellular infiltration.
NTZ Rebound
Discontinuation of NTZ therapy may be necessary for a variety of reasons, including fear of contracting PML after long-term usage, JC virus seroconversion, disease progression despite treatment, pregnancy or the intention to become pregnant, Abs to NTZ, or allergy.35 In these patients, rebound of the primary disease (MS) may be encountered in 22%, with an unusually robust inflammatory response greater than a patient's typical relapse severity before starting NTZ therapy on MR imaging.36
On MR imaging, the appearance of the rebound phenomenon is new enhancing and/or nonenhancing lesions, and the number of these lesions may be greater than in a typical relapse and can be quite severe. Development of enhancement at the margins of old lesions has also been reported.37
Other immunosuppressive mAbs such as efalizumab, rituximab, brentuximab vedotin, alemtuzumab, and eculizumab share with NTZ potential primary and secondary AEs and are discussed in greater detail in the Online Supplemental Data.
Amyloid-Segregating mAbs: Aducanumab
Aducanumab is a human mAb that selectively targets amyloid β aggregates, including soluble oligomers and insoluble fibrils. It is used in the treatment of mild cognitive impairment and mild Alzheimer disease.38 Even with the FDA approval of this drug, there is debate regarding its clinical efficacy in treating mild cognitive impairment and Alzheimer disease, to a certain extent due to the termination of the 2 pivotal Phase 3 clinical trials, 221AD301 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer's Disease (ENGAGE) and 221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer's Disease, following a futility analysis.39 Despite the early termination of both trials, both demonstrated a favorable treatment effect of aducanumab at low doses, but they were discordant at the highest dose because the ENGAGE trial showed no beneficial treatment outcome compared with a placebo, while the EMERGE clinical trial showed a decrease in the rate of cognitive and functional decline. When the final 2 data sets were compared with their respective futility data sets, an improved treatment effect was evident in both studies as additional data were collected. The FDA cited EMERGE as having “substantial evidence of effectiveness to support approval.”40 Nonetheless, at best, the treatment may only slow cognitive and functional decline—raising the question of what pathologies drive continued functional decline as amyloid burden diminishes.
There has been a high rate of imaging-related abnormalities observed in patients treated with Aducanumab. These have been coined amyloid-related imaging abnormalities (ARIA) and occurred in >40% of individuals in the aducanumab 10 mg/kg group.39 Clinically, new signs or symptoms suggestive of ARIA were present in a large number of patients, including headaches in 13%, dizziness in 4%, confusion/altered mental status in 5%, visual disturbance/eye disorders in 2%, and nausea in 2%.39
In September 2022, the American Journal of Neuroradiology published a white paper on ARIA,41 focusing on imaging abnormalities and how to report these. In brief, the current hypothesis of ARIA formation is based on the assumption that amyloid deposition in vessel walls (cerebral amyloid angiopathy [CAA]) may result in loss of vascular integrity and reduced perivascular clearance and may be related to spontaneously occurring microhemorrhages.42 When antiamyloid mAb therapy is initiated, antibody-mediated breakdown of amyloid plaque and mobilization of parenchymal and vascular Aβ increase the load of perivascular drainage.43 This overload of the perivascular clearance pathways, ie, the glymphatic system, transiently increases amyloid deposition in the arterial wall, while at the same time, antibody-mediated inflammation and breakdown of amyloid occur in the vessel wall, all causing loss of vascular integrity and BBB breakdown.44 As a result, proteinaceous fluid and/or red blood cells leak into the parenchyma and/or leptomeningeal space, and this issue results in edema/effusion (ARIA-E) or microhemorrhages/superficial siderosis (ARIA-H). High-risk factors for ARIA are the following: 1) initial treatment period, 2) higher drug doses, 3) ApoE4 genotype—with ApoE4 homozygotes having the highest risk,45 and 4) pretreatment microhemorrhages most consistent with CAA, lobar microhemorrhages, and superficial siderosis.46,47
ARIA-E
The E in ARIA-E stands for edema, effusion, and exudate that may be present, either parenchymal, sulcal, or both. The imaging appearance of parenchymal edema is similar to that of vasogenic edema, ie, absent diffusion restriction, and is best depicted on T2 FLAIR (Fig 4). It occurs mainly in the white matter, with some gray matter involvement. There may be associated local mass effect and gyral swelling. When the leak occurs in the leptomeningeal space, the result is a sulcal effusion or exudate, appreciated on T2 FLAIR sequences within the sulci/subpial spaces.48
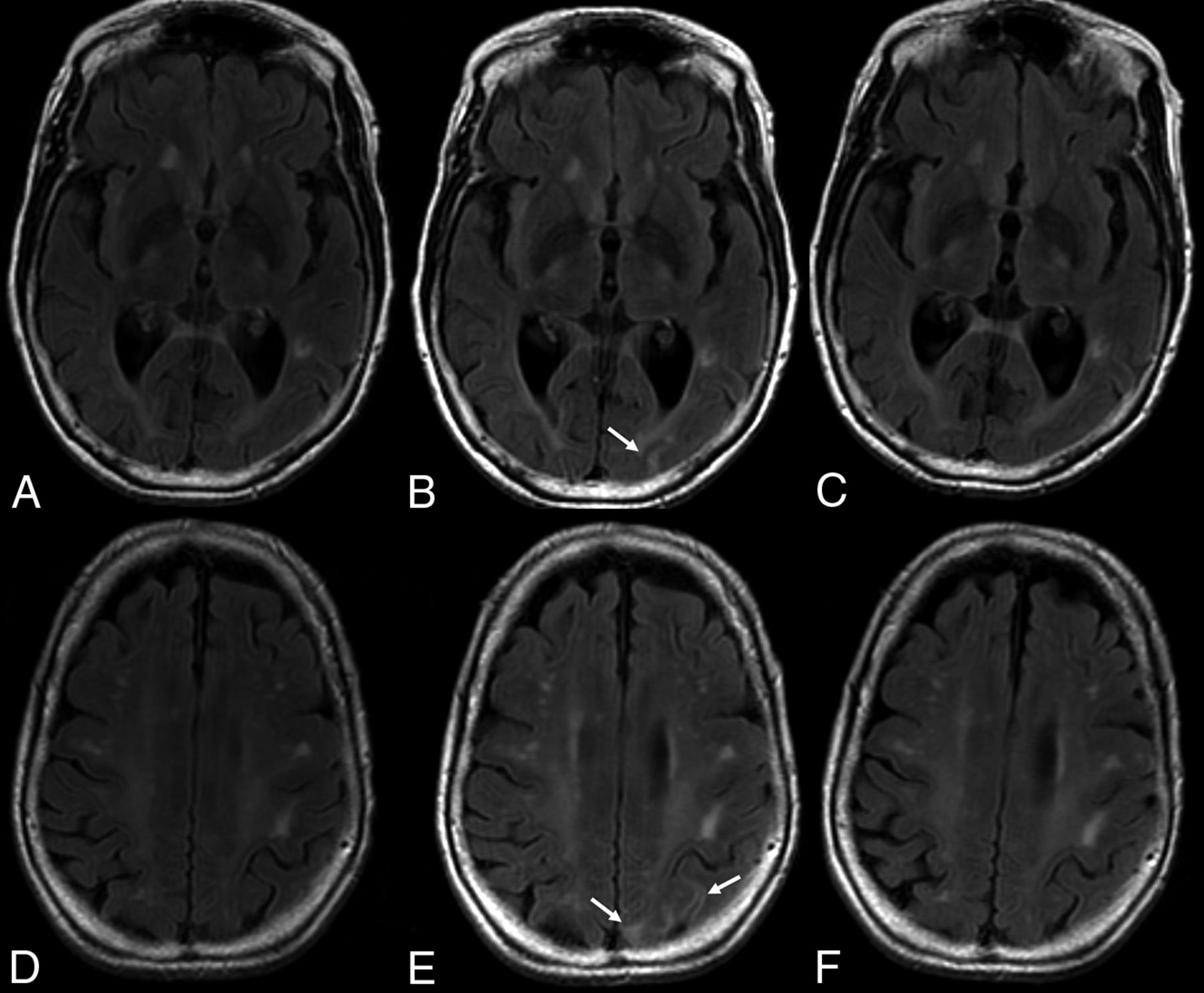
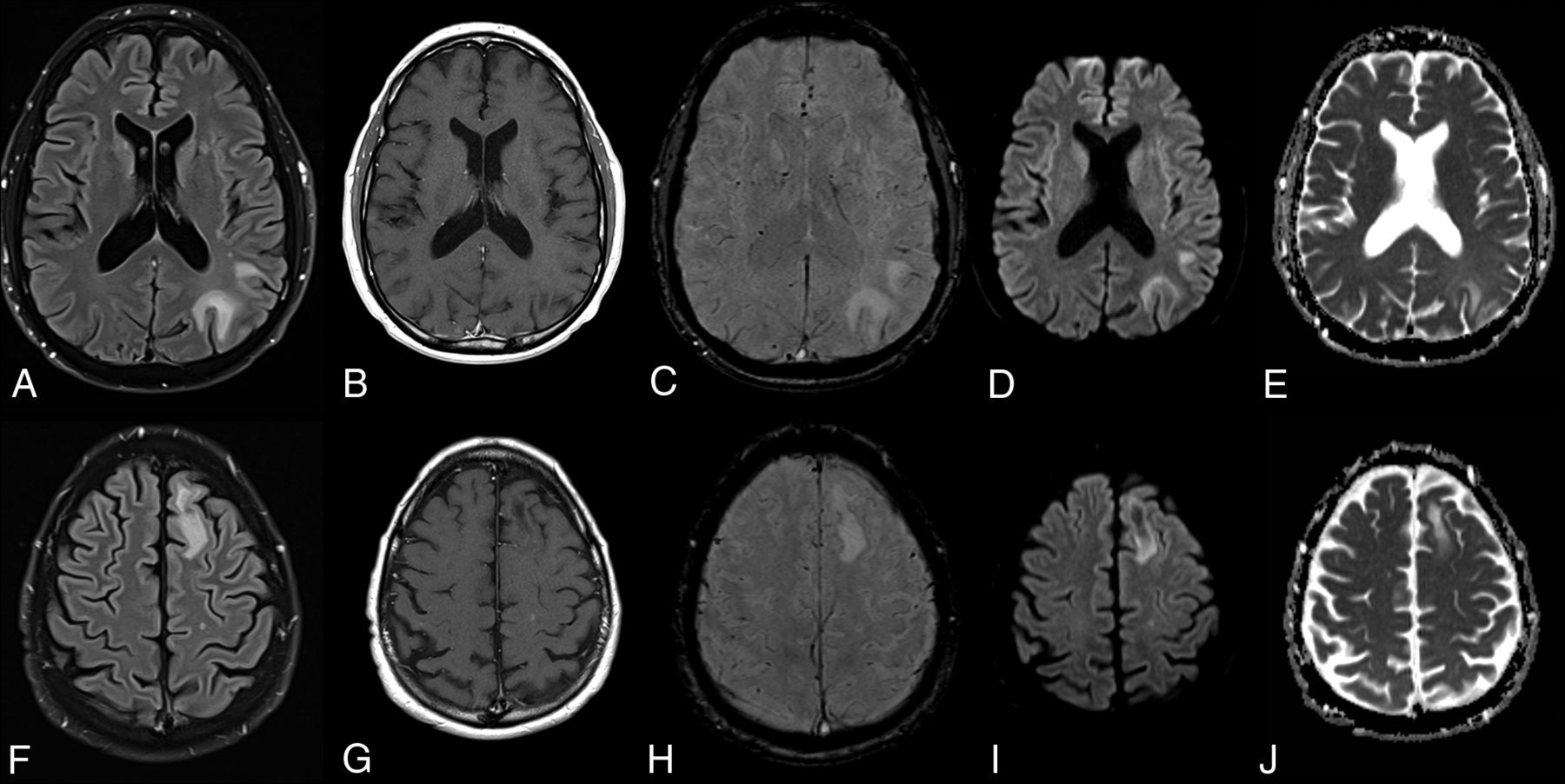
ARIA-E on follow-up. T2-weighted FLAIR scans at baseline (A and D), after 7 months of aducanumab treatment (B and E), and on follow-up 2 months later (C and F) demonstrate, in this 81-year-old patient who remained clinically stable, new development of edematous changes in the left occipital and parietal cortical and subcortical regions (arrows), which spontaneously resolved, in keeping with ARIA-E.
ARIA-E is most often found in dependent (posterior) brain regions—in the following order: occipital, parietal, frontal, and temporal lobes, and, least frequently, the cerebellum. Variable lesion intensity and size ranging from single subtle to multifocal and near-hemispheric46 generally have ill-defined margins and infrequently have circumscribed margins.
ARIA-E is transient and typically persists on MR imaging for about 4–12 weeks, with self-limiting clinical symptoms lasting about 4 weeks after interruption or discontinuation of antiamyloid therapy and has even been reported to resolve under continued dosing.49
Differential diagnoses of ARIA-E include CAA, especially its inflammatory subtype (CAA-RI), PRES, sarcoidosis, or neoplasms, for example, angiocentric lymphoma.
The similarity of ARIA-E to PRES and inflammatory CAA, both on clinical presentation and MR imaging findings, is thought to be related to a similar CNS vascular endothelial dysfunction.46 Differentiating these entities on the basis of imaging alone may, therefore, be difficult, and obtaining the clinical history of treatment with mAbs is crucial in making the diagnosis.
ARIA-H
ARIA-H, particularly in the form of microhemorrhages, is common in untreated, older individuals, and increases in incidence with age. Most cases of ARIA are asymptomatic, with the incidence of symptomatic ARIA in the 10 mg/kg group being 7.5%. Overall, the incidence of ARIA-H was lower than that of ARIA-E, and ApoE4 carriage has an increased risk.39
The H in ARIA-H is hemorrhage, which includes microhemorrhages and superficial siderosis detected on heme-sensitive sequences, ie, T2* gradient recalled-echo and SWI (Fig 5). Intraparenchymal heme leakage results in microhemorrhages that are punctate and rounded, with markedly hypointense foci on T2* sequences, measuring <10 mm in diameter. A leak of heme products into the leptomeningeal or subpial space results in superficial siderosis, which manifests as curvilinear hypointensity along the brain surface.
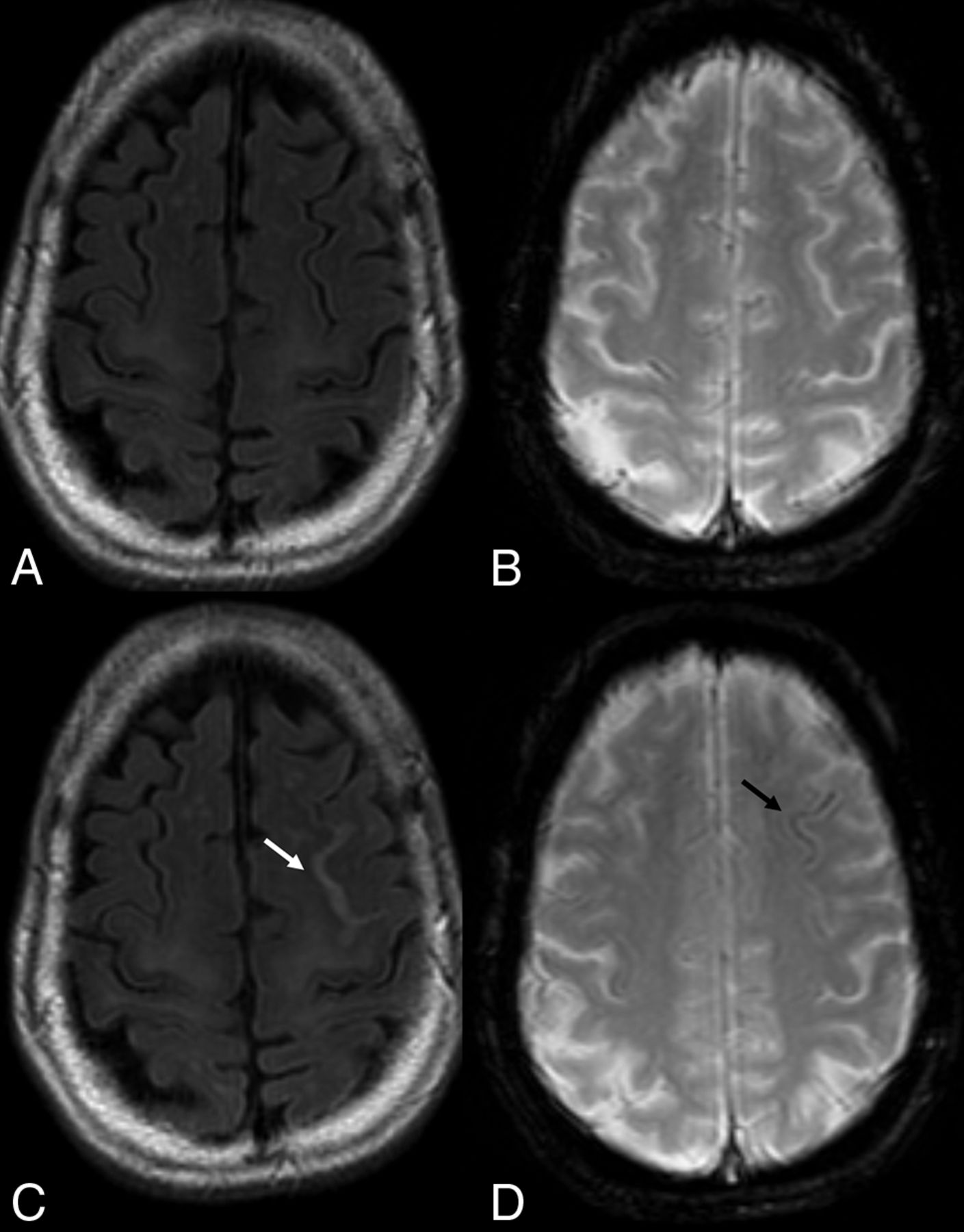
ARIA-H. In this 80-year old woman treated for amnestic mild cognitive impairment with aducanumab, serial imaging demonstrates, in the asymptomatic patient, new foci of blooming artifacts within the left frontal sulcus compared with the baseline scan (A and B; FLAIR and T2 gradient echo sequences). On follow-up 3 months later (C and D), note T2-weighted FLAIR hypersignal surrounding the left superior frontal sulcus (arrow in C), where mild pial siderosis is seen (arrow in D), in keeping with ARIA-H.
Lobar macrohemorrhage (focus of hemorrhage identifiable on T1- or T2-weighted imaging, and usually >10 mm in diameter on gradient recalled-echo) rarely occurs with antiamyloid agents, and when it does, it may be the result of an underlying disease process such as CAA.50 The location of the microbleeds (superficial rather than deep), the type of bleeds (microbleeds as well as siderosis), and their occurrence in conjunction with mAb treatment will help the radiologist differentiate ARIA-H from other causes of microbleeds such as hypertensive angiopathy.
Clinical management is based on patient symptomatology and imaging criteria, with the radiologist playing a major role in the evaluation of patients treated with these new drugs as outlined by the white paper of Cogswell et al41 and summarized in the Online Supplemental Data.
In recent clinical trials, the incidence of ARIA was higher with mAbs that bind the N- (aducanumab, bapineuzemab, gantenerumab) versus C-terminal (ponezumab) and target aggregated-versus-soluble (ponezumab) forms of amyloid β.41 Newer FDA-approved mAbs such as lecanemab that target amyloid β soluble protofibrils appear to have a lower risk of ARIA (approximately 12%).51
Tumor Necrosis Factor–Inhibiting mAbs: Adalimumab
Adalimumab is a human mAb that inhibits tumor necrosis factor-α (TNF-α) by binding to TNF-α and blocking its interaction with surface TNF receptors, thus suppressing inflammation.52 It is used to treat inflammatory conditions including rheumatoid arthritis (RA), psoriasis, psoriatic arthritis, ankylosing spondylitis (AS), inflammatory bowel diseases, juvenile idiopathic arthritis, and hidradenitis suppurativa.52
A variety of immune-mediated AEs have been reported and may manifest clinically as urticaria, psoriasis, lupus-like syndrome, and diabetes mellitus type 1.53 In addition, numerous reports of neurologic AEs, including new development or exacerbation of demyelinating diseases, optic neuritis (Online Supplemental Data), chronic inflammatory demyelinating polyneuropathy, mononeuritis multiplex, Guillain-Barré syndrome, and others have been published.54,55
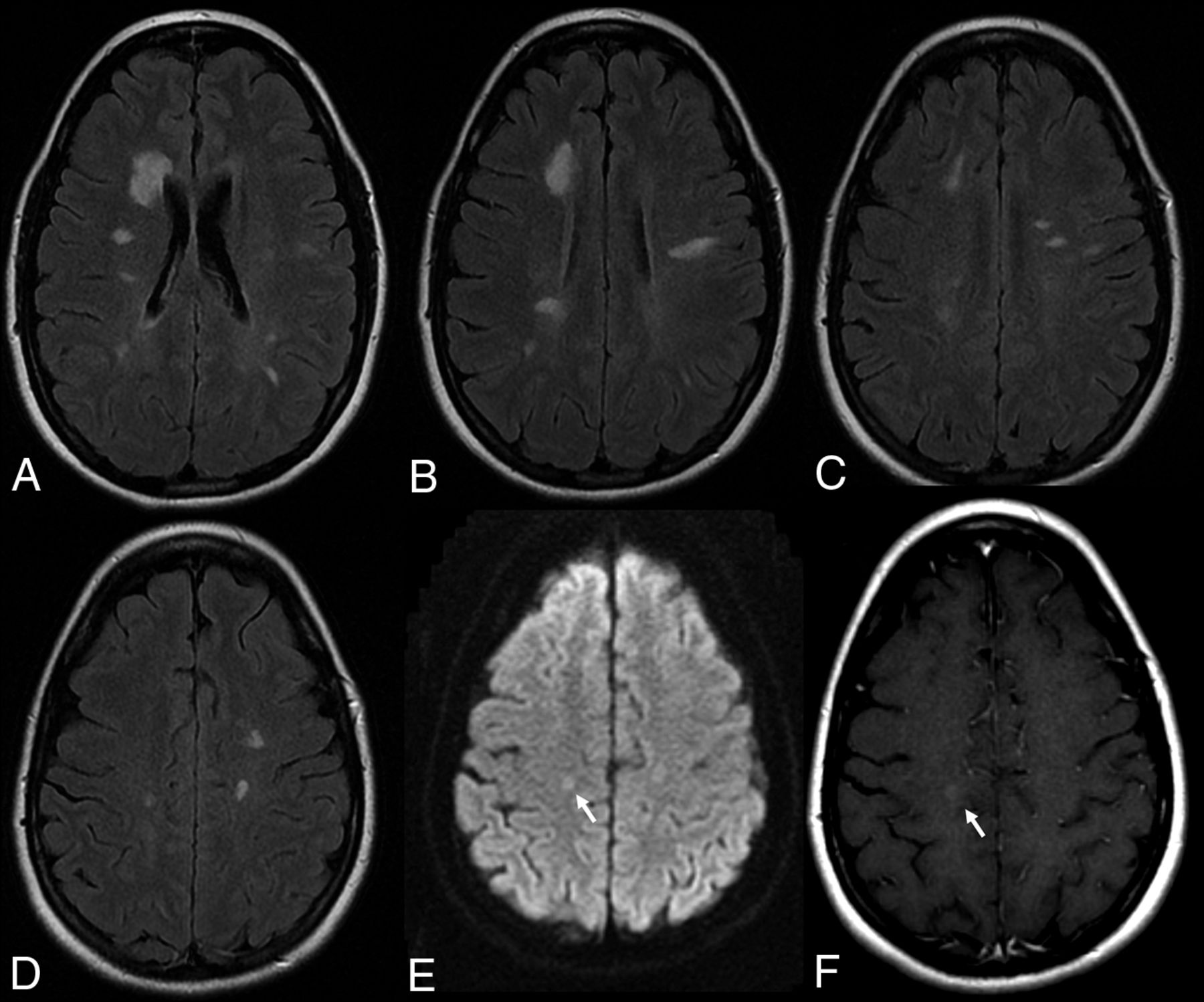
The incidence of demyelinating disease in patients treated with TNF-α inhibitors is estimated to be 0.02%–0.2% of patients receiving this medication (Fig 6).56

Demyelinating lesions after anti-TNF-α AE. This 36-year-old female patient with ankylosing spondylitis was treated with adalimumab and had cognitive decline. MR imaging (A–D: T2 weighted FLAIR, E: DWI, F: Contrast enhanced T1) demonstrates multiple demyelinating plaques with very subtle contrast enhancement (arrow, F).
The relationship between the occurrence of demyelinating disease and TNF-α antagonists is poorly understood, though several theories have been proposed. One hypothesis is that TNF-α antagonists do not penetrate the BBB but peripherally prevent the destruction of autoreactive T cells. Thus, by increasing the number and activity of T cells, more will penetrate into the CNS and increase the autoimmune responses. Another hypothesis states that the inability of TNF-α antagonists to enter the CNS would prevent the inhibition of TNF-α-mediated demyelination in MS. Third, as TNF-α antagonists decrease TNF-α levels systemically but not within the CNS, this could cause an upregulation of TNF-α expression in the CNS, further exacerbating TNF-α-mediated demyelination. Fourth, TNF-α antagonists may inhibit TNF-α-induced interleukin-10 and prostaglandin E2 production, resulting in upregulation of IL-12 and IFN-γ, which are associated with demyelinating disease processes. Fifth, down-regulation of TNF receptor 2 may occur. These receptors are necessary for the proliferation of oligodendrocytes and damage repair. Finally, TNF-α antagonists may unmask a latent infection or neurologic disease, inciting an autoimmune demyelinating process.56 Treatment requires discontinuation of mAb treatment, steroids, and, in severe cases, immunosuppressive drugs.57
CONCLUSIONS
MAbs are currently and will in the future be increasingly used for the management of a wide variety of diseases including neoplastic, autoimmune, and degenerative processes. Knowledge of the type of treatment the patient is undergoing is as important as understanding the potential pathologic mechanism involved in the development of imaging and neurologic sequelae.
While immunostimulating anticancer mAbs such as ipilimumab can lead to proinflammatory conditions such as hypophysitis, immunosuppressive drugs such as NTZ used in a variety of inflammatory conditions and severe MS forms can lead to activation of underlying opportunistic infections (with associated treatment-related IRIS and rebound phenomena). Amyloid-segregating mAbs can lead to amyloid-related imaging abnormalities in which the radiologist plays an instrumental role in patient management. The fourth group of mAbs that is frequently associated with neurologic and neuroradiologic findings is TNF-inhibiting mAbs that cause a higher incidence of demyelinating abnormalities both in the central and peripheral nervous system (Table).
Monoclonal antibody therapies and their use in neurologic diseases
ACKNOWLEDGMENTS
Dr Krings acknowledges the generous support from the Patricia Holt-Hornsby and Dan Andreae Vascular Research Unit and University Medical Imaging Toronto.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received June 19, 2023.
- Accepted after revision July 17, 2023.
- © 2023 by American Journal of Neuroradiology









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.