
Abstract
SUMMARY: Diffuse leptomeningeal glioneuronal tumor is a newly defined entity under the neuronal and mixed neuronal-glial tumors category in the 2016 World Health Organization classification of brain tumors. In this series, we report clinical, radiologic, and histologic findings in 7 cases of diffuse leptomeningeal glioneuronal tumor. Our cases and literature review indicate that the most characteristic imaging finding is diffuse intracranial and intraspinal nodular leptomeningeal thickening and enhancement. This is often associated with small cyst-like, nonenhancing lesions. It should be noted that tumors sometimes bear nontypical features, for example, presenting as a solitary spinal cord mass without leptomeningeal involvement or with a dominant intracranial mass. In children with characteristic imaging findings and without clinical features of infection, the radiologist has an opportunity to promptly raise the possibility of diffuse leptomeningeal glioneuronal tumor, and thereby, affect streamlined diagnostic evaluation.
ABBREVIATIONS:
- DL-GNT
- diffuse leptomeningeal glioneuronal tumor
- WHO
- World Health Organization
Diffuse leptomeningeal glioneuronal tumor (DL-GNT) is a newly defined neuronal and mixed neuronal-glial tumor described in the 2016 World Health Organization (WHO) classification of brain tumors.1 This entity has previously been described as disseminated oligodendroglial-like leptomeningeal tumor, dysembryoplastic neuroepithelial tumor-like neoplasm, meningeal gliomatosis, diffuse leptomeningeal neurocytoma, diffuse leptomeningeal gangliocytoma, and diffuse leptomeningeal oligodendrogliomatosis.2
DL-GNT is a rare neoplasm that is more common in male children. Adult cases are rare.1 The exact incidence is unknown, as only limited cases have been reported in the published literature. Most of these tumors show slow progression and have been managed by surgical resection, radiation therapy, and/or chemotherapy. Given the rarity of this tumor, an optimal evidence-based management strategy has yet to be developed.
Predominant diffuse abnormal nodular leptomeningeal growth without evidence of a primary intraparenchymal focus is the most common imaging feature.3,4 Characteristic diffuse abnormal leptomeningeal enhancement is most commonly seen in the basal cisterns and posterior fossa, and along the spinal cord. Small, nodular T2-hyperintense leptomeningeal lesions resembling cysts are sometimes seen. Though discrete intraparenchymal lesions are unusual, they have been most commonly described in the spinal cord.4,5 The wide spectrum of histologic, radiologic, and clinical features often mimicking chronic infection or leptomeningeal carcinomatosis, along with very low incidence rate can make prospective tumor identification by imaging challenging.1⇓⇓-4
Here, we report the clinical characteristics, imaging features, and histopathologic findings of DL-GNT in 7 patients with emphasis on imaging in hopes of increasing awareness of this entity and facilitating rapid diagnosis in affected patients.
CASE SERIES
Institutional review board approval was waived and not required for this retrospective case series. Seven cases were identified from 3 different institutions: Vanderbilt University Medical Center in Nashville; Great Ormond Street Hospital in London; and McGovern Medical School, The University of Texas Health Science Center at Houston between October 2015 and December 2019.
Electronic medical record review yielded age, clinical presentation, and histopathologic diagnosis. All patients underwent brain and spinal cord imaging at either 1.5 or 3T with the following key sequences obtained in all cases: T2-weighted, FLAIR, postgadolinium T1-weighted, and diffusion-weighted. All MR imaging studies were reviewed independently by 2 fellowship-trained pediatric neuroradiologists (S.P., A.S.).
Clinical data are summarized in On-line Table 1. All patients were male. Patients presented between 3 and 14 years of age with chronic headaches (n = 4), new onset seizure (n = 2), and gait imbalance (n = 1). Detailed infectious work-up including CSF analysis for infection was negative in all patients. All 7 patients underwent biopsy.
On MR imaging, diffuse nodular intracranial leptomeningeal thickening was present in 5 of 7 (72%) cases (On-line Table 1, Fig 1A, -B). One of the 5 (20%) cases had infratentorial predominance, while the other 4 (57%) cases demonstrated diffuse intracranial involvement with all demonstrating basilar cisterns involvement. Of note, the remaining 2 (29%) cases showed minimal to no intracranial leptomeningeal disease. Multiple intracranial lesions that did not enhance or suppress on FLAIR (“cystic-appearing”) were noted in 2 of 7 (29%) cases (On-line Table 1, Fig 2A). Hydrocephalus was noted in 5 of 7 (71%) cases. A dominant intracranial parenchymal mass lesion was noted in 1 of 7 (15%) cases (On-line Table 1, Fig. 3A-C).
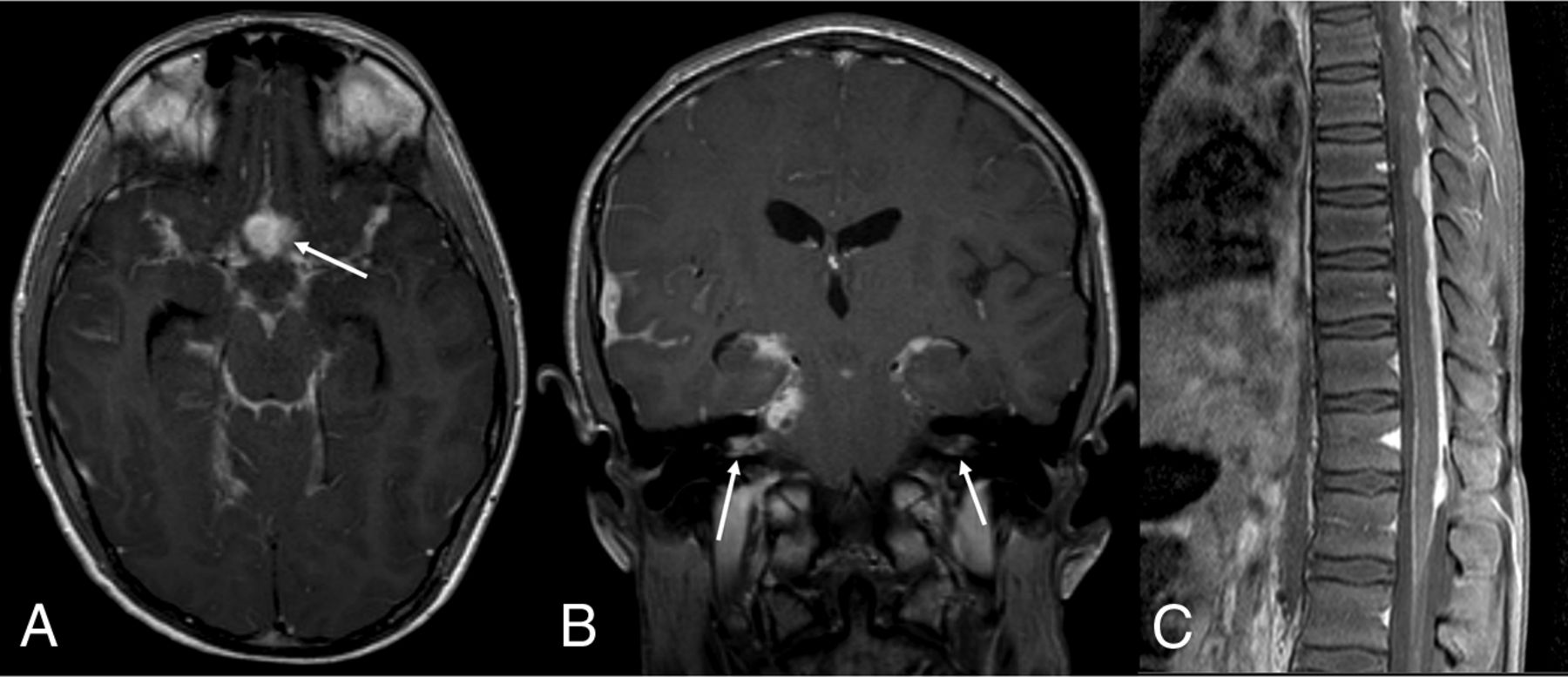
DL-GNT with diffuse leptomeningeal disease. Axial and coronal postcontrast T1-weighted (A and B) MR images demonstrate diffuse intracranial nodular/mass-like leptomeningeal enhancement. This is most prominent within the basilar cisterns, with masslike enhancement at the level of the optic chiasm (arrow). Also seen is abnormal enhancement extending along both cranial nerve VII/VIII complexes (arrows). Abnormal enhancement extending along cranial nerves is not uncommon in this entity. Sagittal T1-weighted postcontrast fat-suppressed image of the thoracic spine (C) shows thick, nodular leptomeningeal enhancement predominantly along the dorsal thoracic cord. The constellation of imaging findings seen in this case is the most commonly described in DL-GNT.
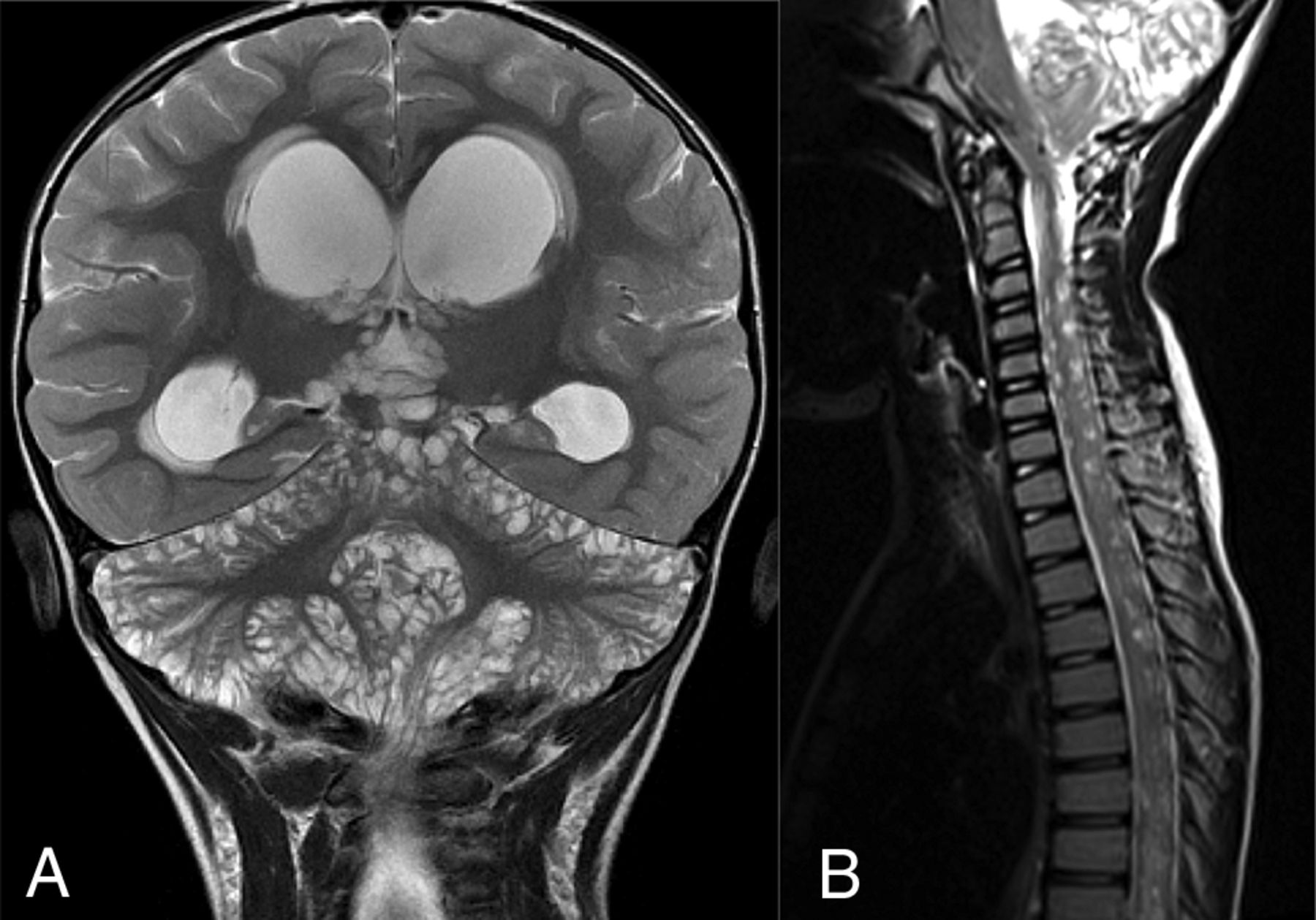
DL-GNT with diffuse “cyst-like” foci of T2 prolongation involving the brain and spine. Coronal T2-weighted image of the brain (A) demonstrates numerous foci of T2 prolongation located predominantly along the leptomeningeal surfaces of the cerebellum. These did not enhance after contrast administration or suppress on FLAIR (not shown). Also seen is hydrocephalus with transependymal edema as evidenced by abnormal periventricular T2 prolongation. Sagittal T2-weighted image of the cervicothoracic spine (B) in a different patient shows numerous small cervicothoracic intramedullary cyst-like lesions expanding the spinal cord. These cyst-like lesions are very suggestive of DL-GNT.

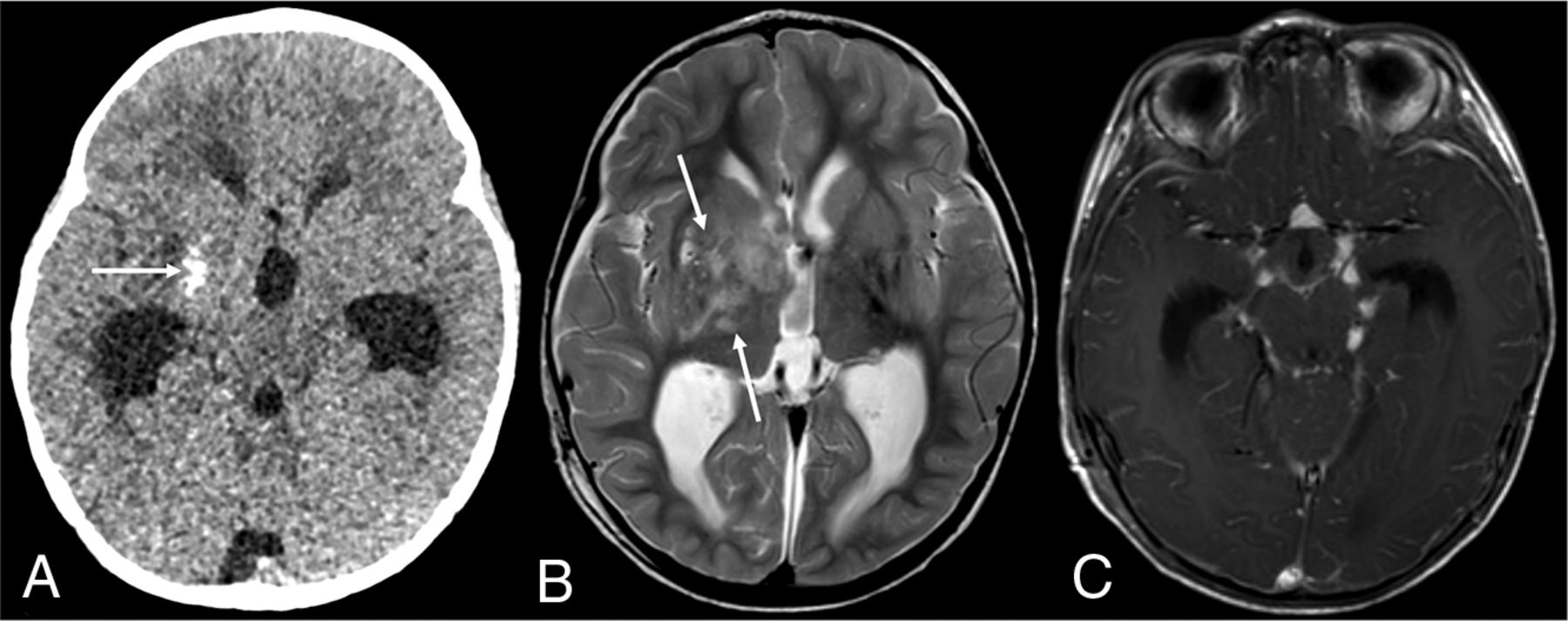
DL-GNT with dominant intracranial parenchymal mass. Axial noncontrast head CT in a patient with findings of hydrocephalus (A) shows ill-defined calcification within the right basal ganglia (arrow). Basal ganglia germinoma was initially considered in the differential diagnosis. Axial T2-weighted MR imaging (B) shows an ill-defined, slightly expansile area of T2 prolongation within the right putamen, thalamus, and caudate head overlapping with the calcified region demonstrated on CT (arrows). Minimal enhancement was seen in the right anterior thalamus (not shown). Axial T1-weighted postcontrast image (C) shows numerous foci of nodular supratentorial leptomeningeal enhancement lining the basilar cisterns. Despite the atypical-appearing mass involving central gray matter, the presence of characteristic diffuse supra- and infratentorial nodular deposits led to prospective consideration of DL-GNT in the differential diagnosis.
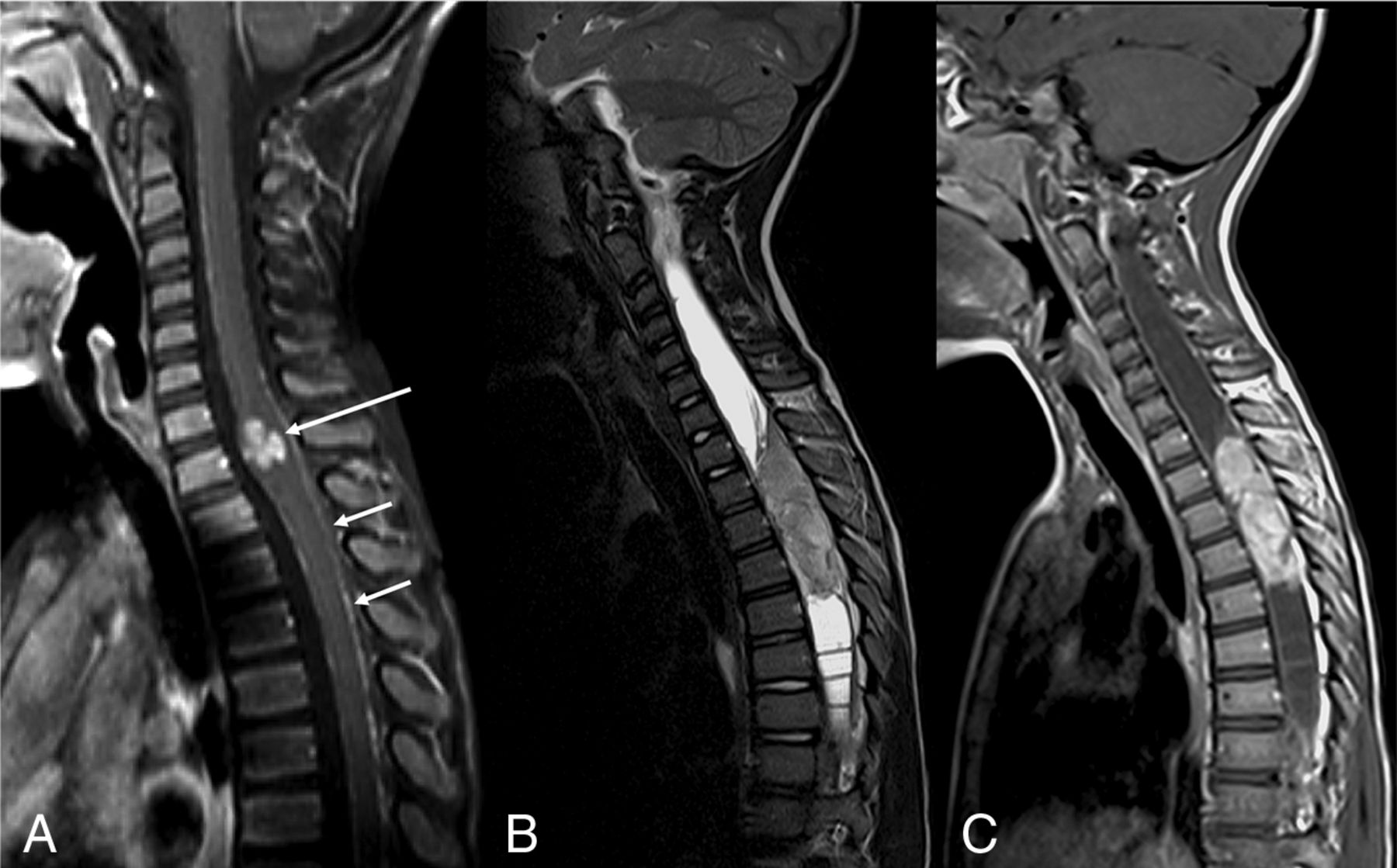
The patterns of spinal involvement included: diffuse, dorsal-predominant nodular leptomeningeal thickening (On-line Table 1, Fig 1C) in 5 of 7 (72%) cases, intramedullary spinal cord mass (On-line Table 1, Fig. 4A-C) in 2 of 7 (29%) cases, and multifocal cystic spinal cord lesions (On-line Table 1, Fig 2B) in 2 of 7 (29%) cases. Interestingly, 1 patient with cystic-appearing spinal cord lesions and another patient with a large intramedullary mass were the ones without associated nodular leptomeningeal spinal disease.
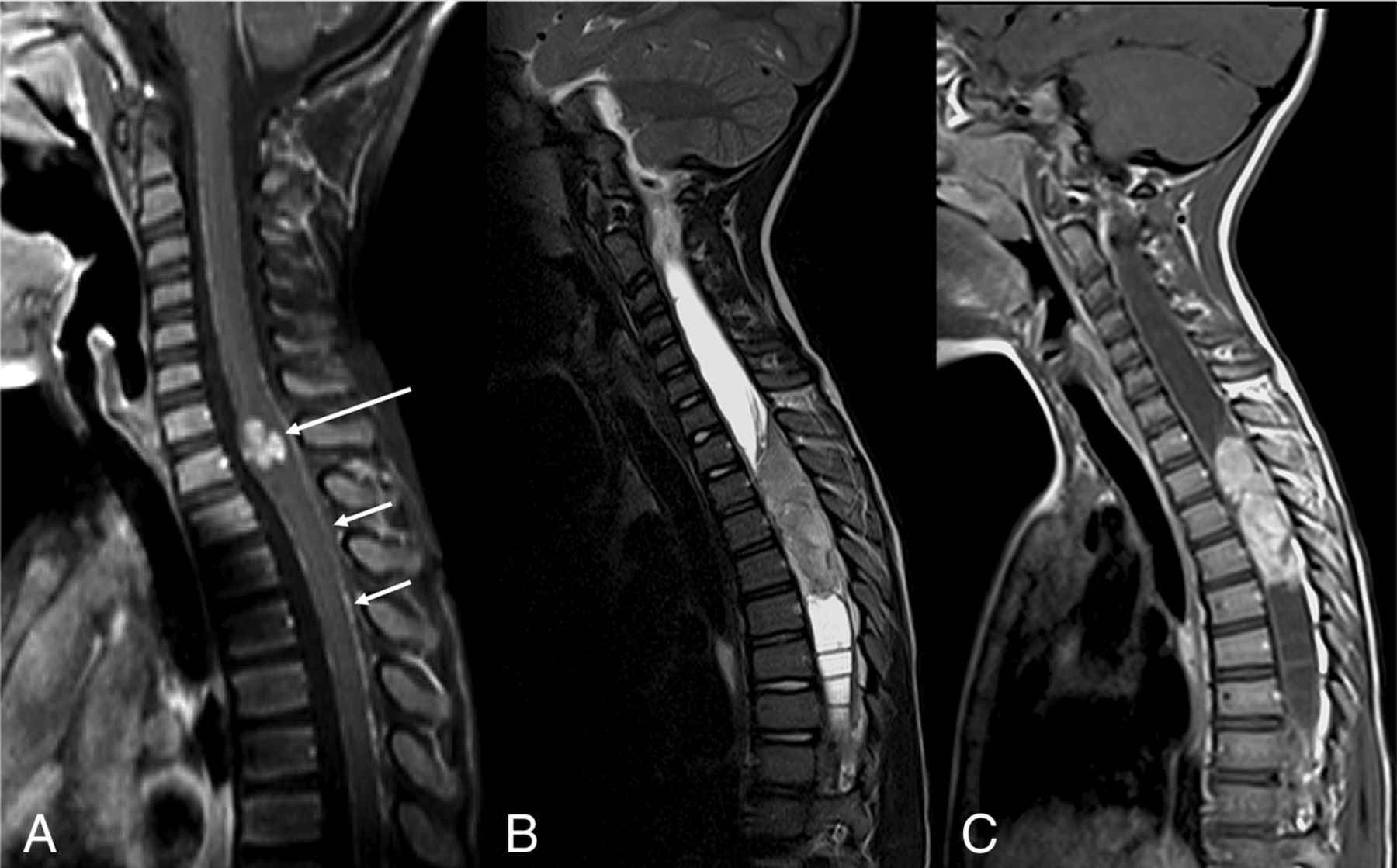
DL-GNT with isolated spinal cord mass. Sagittal T1-weighted postcontrast image of the cervical spine (A) demonstrates a small focal enhancing mass (long arrow) expanding the lower cervical spinal cord. Scattered areas of linear leptomeningeal enhancement are also seen along the dorsal (short arrows) and ventral aspects of the cord. Sagittal T2-weighted and T1-weighted postcontrast images of the spine (B and C) in another patient demonstrate a large hypointense, enhancing intramedullary mass extending from T2 to T5, with associated large, septated syrinx. These 2 examples highlight that intramedullary masses in DL-GNT can exhibit different appearances.
All the cases underwent biopsy and histopathologic analysis. Biopsy samples were obtained from cystic lesions in 2 of 7 (29%) cases, isolated spinal cord masses in 2 of 7 (29%) cases, and leptomeningeal nodular lesions in 2 of 7 (29%) cases. Histopathology demonstrated strong reactivity for OLIG2, MAP2, and S-100, with variable expression of glial fibrillary acidic protein and synaptophysin. BRAF fusion was noted in 2 of 7 (29%) cases, and deletion of chromosome arm 1p was noted in 2 of 7 (29%) cases. We did not see co-deletion of chromosome 1p19q in any cases (On-line Table 1).
DISCUSSION
This series describes the clinical, pathologic, and radiologic features in 7 cases of DL-GNT. Most existing publications on this topic have been presented in the pathology and oncology literature, and we believe that this imaging-focused series is the largest in the radiology literature to date. This is important because many tumors previously classified otherwise (eg, disseminated oligodendroglial-like leptomeningeal tumor) are now known to represent DL-GNT, and this tumor may not be as rare as once suspected. Furthermore, despite its somewhat distinctive imaging presentation, the diagnosis frequently remains unsuspected early in the patient’s clinical course, sometimes resulting in excessive diagnostic testing for other entities with increased cost and delayed diagnosis. In expeditiously raising suspicion for DL-GNT when the characteristic appearance is encountered in a patient without clinical findings of infection, the radiologist has an opportunity to facilitate rapid biopsy, diagnosis, and treatment of affected patients.
Histopathologically, tumor consists of 2 cell populations, neuronal and glial. The neuronal component consists of synaptophysin-positive neurocytes, while the glial component features glial fibrillary acidic protein–positive astrocytes.4 Histologically, DL-GNTs are low- to moderate-cellularity lesions consisting of relatively monomorphous oligodendrocyte-like cells with a “glioneural commitment,” embedded in a desmoplastic or myxoid leptomeningeal stroma. Immunohistochemical features of the tumor cells include strong reactivity for OLIG2, MAP2, and S-100, with variable expression of glial fibrillary acidic protein and synaptophysin. Tumors commonly harbor BRAF fusions as well as deletions of chromosome arm 1p, either alone or occasionally combined with 19q deletion (hence previous use of the descriptor “oligodendroglioma-like”). Immunostaining is negative for NeuN, epithelial membrane antigen, and mutant IDH1, distinguishing DL-GNT from adult oligodendrogliomas (R132H).1⇓⇓-4 In summary, histopathologically, DL-GNTs are low-grade neoplasms with primarily oligodendroglioma-like features, neuronal differentiating capacity, and tendency toward extensive leptomeningeal dissemination.
Based on the literature review of published DL-GNT cases, the median age at presentation is approximately 4 years and there is a male predilection (58 of 91) (64%).2,6⇓-8 In our case series, all patients were male, with a median age of 6 years.
On-line Table 2 summarizes features of cases previously reported in the literature. Our case series was similar to those reported in the literature in terms of percentage of cases with diffuse intracranial nodular leptomeningeal thickening (72% in our series, 73% in the literature); diffuse intraspinal nodular leptomeningeal thickening (72% of cases, 62% in the literature); and intramedullary spinal cord mass (29% of cases, 20% in the literature). Our series had a smaller number of cases with multiple intracranial nonenhancing lesions with T2 prolongation (29% in our series, 43% in the literature); and more cases with hydrocephalus (71% in our series, 29% in the literature).
Although extensive intracranial leptomeningeal tumor has been considered a defining feature of DL-GNT, 29% of the cases in our series demonstrated minimal to no obvious intracranial leptomeningeal involvement (compared with 19% in the literature). Compared with previously reported studies, our series had a smaller percentage of cases with multifocal nonenhancing intramedullary spinal cord lesions with T2 prolongation (29% of cases, 43% in the literature). A single case in our series was associated with extensive intracranial and spinal leptomeningeal tumor deposits with an ill-defined, minimally-enhancing, calcified mass involving the right putamen, thalamus, and caudate nucleus; a similar finding has previously been reported by Gardiman et al.9
The imaging presentation of DL-GNT is not well-understood. The key MR imaging findings of DL-GNT based on our case series and literature review include diffuse leptomeningeal thickening and multifocal cystic-appearing lesions in both the brain and spinal cord. These findings may be present in conjunction with or without isolated spinal cord mass.
Based on our literature review, DL-GNT was frequently misdiagnosed as tuberculosis, especially in cases from Europe and the Asia-Pacific region.10 The broad imaging differential diagnosis includes infection (eg, bacterial or tuberculous meningitis, neurocysticercosis), other CNS tumors including disseminated high-grade and low-grade neoplasms (eg, atypical teratoid/rhabdoid tumor, high-grade glioma, primary diffuse leptomeningeal gliomatosis, medulloblastoma, pilomyxoid astrocytoma), leukemia or leptomeningeal lymphomatosis, phakomatosis (eg, neurocutaneous melanosis, Sturge-Weber syndrome), and neurosarcoidosis.
Correlation with clinical features of infection and evaluation of the character of leptomeningeal involvement can help to differentiate DL-GNT from infection. Bacterial meningitis tends to have smooth rather than nodular sulcal-cisternal enhancement, and tuberculous meningitis more often causes confluent basilar enhancement. The vesicular stage of neurocysticercosis may present with multiple cysts with internal characteristic scolex “dots” deep within deep sulci, with intense inflammatory reaction in the adjacent parenchyma, that is not typical of DL-GNT.
Apart from noninfectious CSF findings, the key in differentiating DL-GNT from other entities is identification of diffuse leptomeningeal involvement of the brain and spinal cord, with or without multifocal cystic-appearing changes, or the presence of a solitary solid spinal mass.
CONCLUSIONS
Given the recent changes in classification by the WHO, as well as the rarity of this neoplasm, many radiologists may not yet be aware of DL-GNT. Furthermore, this condition may be unexpected by referring clinicians caring for patients with a newly discovered leptomeningeal disease process. In recognizing the various imaging patterns illustrated in this series, the radiologist may be the first member of the clinical team to suspect this rare diagnosis.
There should be a high index of suspicion for DL-GNT when the characteristic findings are observed in the absence of clinical and laboratory signs of infection, and when a dominant neural axis mass is absent or disproportionately small or indolent-appearing relative to the marked degree of leptomeningeal disease. While these imaging findings are typical, it should be noted that DL-GNT can also present as a solitary spinal cord mass without leptomeningeal involvement. When the diagnosis is suspected, prompt biopsy should be performed to reduce the time to definitive diagnosis and appropriate treatment.
Footnotes
Previously presented in part at: Annual Meeting of the American Society of Pediatric Neuroradiology 2019, January 18–21; Los Angeles, California.
Disclosures: Kshitij Mankad—UNRELATED: Expert Testimony: Medicolegal reporting in UK; Payment for Lectures Including Service on Speakers Bureaus: Speaker honoraria – Novartis, Siemens. Asha Sarma—UNRELATED: Royalties: Cambridge University Press, Comments: Unrelated textbook.
References
- Received April 8, 2020.
- Accepted after revision June 1, 2020.
- © 2020 by American Journal of Neuroradiology








{kind=link}
{kind=link}
{kind=link}
{kind=link}