
Abstract
Summary: Increased propensity for tumor formation in neurofibromatosis and tuberous sclerosis exists because of defective tumor-suppressor genes. Although different tumor-suppressor genes may be involved in neurofibromatosis and tuberous sclerosis, at the cellular level these genes share rather common enzymatic pathways. We believe these genetic malfunctions have resulted in a cumulative or additive effect for rapid growth of optic glioma in the following unusual case that has hybrid phakomatosis.
Optic glioma is commonly seen in the setting of neurofibromatosis type 1 (NF-1) and has a favorable course in childhood due to relatively benign disease (1, 2); however, coexistence of more than one phakomatosis such as neurofibromatosis and tuberous sclerosis (TS) can augment the phenotypic expression, which in our case is optic glioma (OG), because of additive or synergistic effect at the genetic level. Recent evidence indicates that formation of neoplasm in both conditions can be due to deficiency of tumor-suppressor genes. In this article, we present a case of an infant with combined NF-1 and TS who had a worsening course due to rapidly growing OG.
Case Report
The infant has history of maternal NF-1 and paternal TS. Her sister has NF-1, and the father’s mother and sister have TS. The patient was delivered by cesarean section at 39 weeks because of breech presentation. Otherwise, her pre- and perinatal course was unremarkable. At birth, her physical examination was significant for café-au-lait spots, hypopigmented macules, and a 3–4-cm Shagreen patch on the back of her right thigh. She was normocephalic with soft and flat fontanel. Ear pits were prominent, but otherwise the auriculocular structures were symmetric and normal in position. She had normal genitalia. The kidneys were normal at sonography. Her back was straight. Soon after birth, she developed right-sided proptosis. The cornea and pupil were also larger on the right, although the pupillary reflexes were preserved. Both optic nerves were normal on fundoscopic examination. The ocular pressures were also normal. MR imaging of brain at age 3 months revealed multiple subependymal nodules, T1 hyperintensities in the white matter, and a left foramina Monro lesion. The myelination pattern was normal. Orbits were assessed with a dedicated MR imaging study. There was right-sided proptosis, but the optic nerves were symmetric without any discrete enhancing mass. At 4 months of age, she was able to hold her head up, make eye contact, follow, smile, and blow bubbles. She was rolling over and starting to crawl at 5 months old.
The patient returned to the radiology department at age 8 months for a follow-up MR imaging study. The multiple subependymal nodules and left foramina Monro nodule were found to be stable. The white matter T1 hyperintensities appeared more conspicuous, with two new foci of similar signal intensity abnormalities being seen in the left superior frontal region.
Normal progression of myelination was present. The right optic nerve was now enlarged with intense enhancement (Fig 1). The appearance was consistent with developing OG. The neuroophthalmology team chose to follow the patient closely as per the family request. Nonetheless, the OG continued to show rapid growth and enhancement with a worsening proptosis (Figs 2 and 3). This led to the institution of chemotherapy at age 2 years with gradual response of the tumor (not shown).

Coronal T1-weighted images (TR/TE: 523/14) with contrast enhancement and fat suppression and obtained through the orbits reveal an enhancing right-sided OG at age 8 months.
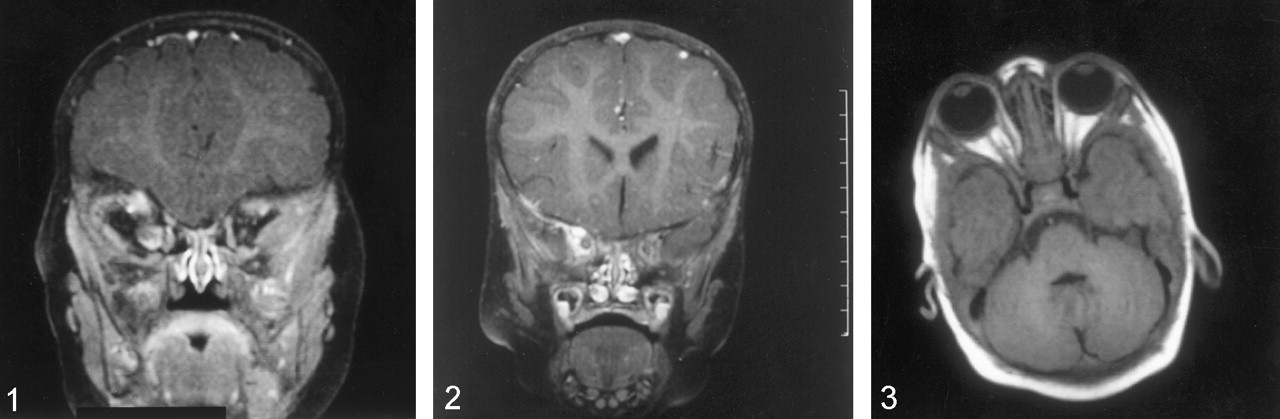
Coronal T1-weighted images (TR/TE: 523/14) with contrast enhancement and fat suppression and obtained through the orbits demonstrate interval worsening of right-sided OG at age 18 months.

Axial postcontrast T1-weighted images (TR/TE: 670/14) with fat saturation demonstrate marked worsening of right-sided proptosis due to rapid growth of the right OG at age 23 months).
Discussion
NF-1 and TS are neuroectodermal dysplasias, namely, phakomatosis with characteristic lesions and multiple clinical stigmata. To the best of our knowledge, there have been only eight cases reported in the literature (3–5) in which both conditions have occurred simultaneously in the same patient. Ours is the ninth such case described. For the purpose of this article, we named the coexistence of these dysplasias in the same patient “hybrid phakomatosis.” Although our patient did not undergo genetic testing, separate diagnosis for NF-1 and TS was established by family background, as well as specific clinical and imaging criteria as specified elsewhere (6, 7).
Some light has recently been shed on the genetic basis of phakomatosis. The determination of a full genetic profile has been hindered by several factors. The NF-1 gene has increased susceptibility to spontaneous mutation (8) that is, in turn, responsible for most of these cases. The patients can be homo- or heterozygotic. In addition, NF-1 has a variable expression, possibly because of genetic mosaics, making it very hard to determine which patients will develop the disease or its severe complications (9). Within the past several years, however, the NF-1 gene has been identified on chromosome segment 17q11.2. The NF-1 gene encodes a protein called “neurofibromin.” The recent evidence from animal research focused on this gene indicates that, among its many roles, the most important function can be the tumor suppression through the cellular GTPase systems. The homozygotic deletion can accelerate the tumor formation in patients with NF-1, especially in plexiform neurofibroma and malignant tumor (10). Excellent review of the subject has been provided in the literature (11).
There is also growing evidence in the literature indicating a similar genetic basis for TS-induced neoplasms. The recently mapped TSC1 and TSC2 genes are believed to function as tumor suppressors through the same cellular systems. Complete inactivation of TSC2 gene has been implicated in the formation of hamartomas and possibly renal cell carcinomas (12, 13).
Approximately 15–20% of patients with NF-1 will develop OG, sometimes as the first sign of subtle NF-1. In Lund and Skovby’s series (14), the mean age of diagnosis was 6.4 years, with the average of duration of visual symptoms before diagnosis being 2 years. MR imaging has been shown to be far more superior to visual field testing, fundoscopy, and visually evoked potentials to detect these lesions (15). Therefore, routine cranial imaging is now recommended to exclude OG in patients with NF-1 in the first few years of life (16). OG has a favorable histologic course and slow growth in NF-1 compared with OG of the non-NF-1 variety (1), with most of these being pilocytic astrocytomas. It has been reported in a long-term follow-up of more than 20 years that tumor growth and the difference between treated and untreated lesions can be hardly noticeable (2). Aggressive malignant behavior is seen less than 20% of patients (17), especially for tumors detected in infancy (18). It has been hypothesized that, in patients with NF-1, there are fundamental pathophysiologic differences between patients with and those without OG (19). A striking association has been observed between the OG and the other CNS neoplasms, independent of age.
Nonetheless, it has been reported that patients with NF-1 who have had prior negative findings for OG are still at risk for forming OG later in life. In a series of neurofibromatosis-related OGs, Massry et al (20) described two cases among seven for which prior neuroimaging findings were negative. They suggested that prior normal CT or MR findings of orbits do not exclude the future development of OG, because slightly older cases took much longer (ie, 2–3 years) to form OG. Conversely, a 15-mm OG in our case had developed only 8 months after initial imaging (Fig 1).
Although the TS and NF1 genes are not located closely at the genetic locus, their functions at the cellular level are intimately related. They both suppress the oncogenes through the GTPase systems (21). We believe this might have resulted in additive or synergistic effect of dysfunctional tumor-suppressor genes in rapid formation of OG in this unusual patient.
Conclusion
We report a rare occurrence of neurofibromatosis and tuberosclerosis in the same patient. These are genetically transmitted disorders. Their phenotypic expression reflects underlying dysfunction of specific genes, also known as tumor suppressor genes. At the cellular level, tumor suppressor genes may share the same enzymatic pathways. We believe their additive or synergistic effects in our patient resulted in an aggressive course of a rapidly developing infantile optic glioma.
References
- Received December 18, 2002.
- Accepted after revision April 14, 2003.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.