
Abstract
Summary: Encephalocraniocutaneous lipomatosis (ECCL) is a rare neurocutaneous syndrome characterized by unilateral scalp, facial, and ocular lesions and ipsilateral cerebral malformations. To define the neuroimaging features of this disorder we studied two patients affected by ECCL and compared our data with those reported in the literature. Sonographic, CT, and MR imaging examinations showed quite specific CNS findings that are highly suggestive of the diagnosis of ECCL. To our knowledge this is the first report of a complete neuroradiologic evaluation and follow-up of this disorder.
Encephalocraniocutaneous lipomatosis (ECCL) is a rare neurocutaneous syndrome first described in 1970 by Haberland and Perou (1); it is characterized by unilateral lipomas of the cranium, face, and neck, ipsilateral lipodermoids of the eye, and ipsilateral brain anomalies (1, 2). We report the results of a complete neuroradiologic evaluation and follow-up of two children affected by ECCL in order to document the sonographic, CT, and MR features of this syndrome.
Case Reports
The transfontanellar sonographic studies were obtained on a 7-MHz sectorial array; the MR studies were obtained on a 1.5-T system with a spin-echo sequence.
Case 1
A male infant of healthy, unrelated parents was born at term after an uncomplicated delivery. At birth, he was noted to have ocular alterations on the right side, a nodular lesion on the upper eyelid, a dermoid of the scleral limbus, epicanthus inversus, hypertelorism, and ipsilateral cutaneous lesions of the head and face with alopecia. A skin biopsy showed fibro-fatty tissue. Results of a neurologic examination at birth were normal. At 4 months of age, the head circumference was in the 97th percentile, and mild left-sided hemiparesis and moderate psychomotor delay were noted. At age 3 years, the patient developed episodes of simple absences, and over the following years developed severe epilepsy. At age 7, slight hemiparesis and mild mental retardation were still present. The cutaneous and ocular lesions remained unchanged over time.
Sonographic, CT, and MR studies, performed at age 2 months, showed enlargement of the lateral ventricle on the right side, mainly at the occipital horn, and of the sylvian fissure, with incomplete insular opercularization (Fig 1A); a middle cranial fossa arachnoid cyst; a cerebellopontine angle lipoma (Fig 1B); and a small right cerebral peduncle, probably a consequence of the reduction in the right corticospinal fibers. CT scans also showed right frontal corticopial calcifications.
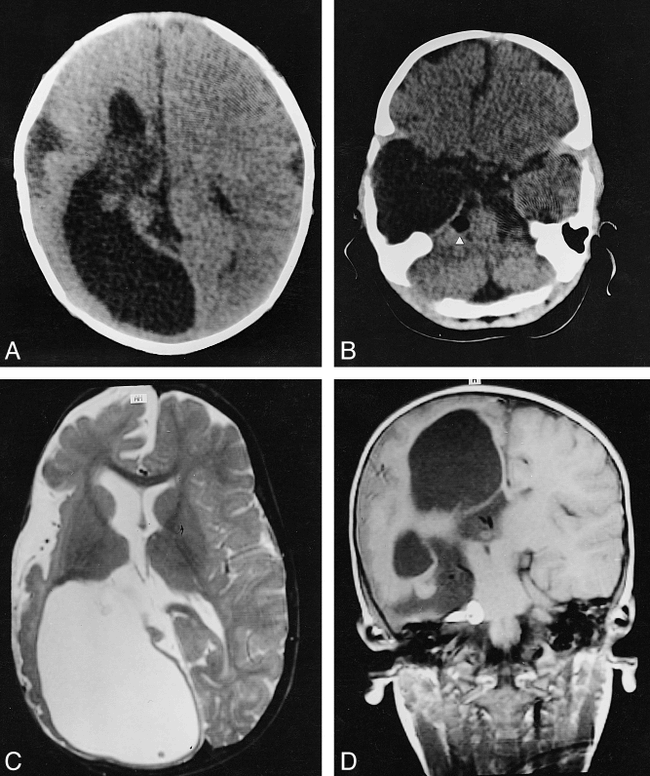
Case 1.
A and B, CT scans obtained at 20 days of life show an enlargement of the right lateral ventricle, mainly at the occipital horn, and of the right sylvian fissure, with incomplete opercularization of the insula (A), and a large right middle cranial fossa arachnoid cyst and a cerebellopontine angle lipoma on the same side (arrowhead, B).
C and D, MR images obtained at 19 months of age. Axial T2-weighted image (3000/120/1 [TR/TE/excitations]) (C) confirms the enlargement of the right lateral ventricle and the abnormal opercularization of the insula; the right hemispheric cortex appears thick, with few and irregular sulci. Coronal T1-weighted image (510/15/2) (D) shows the right pontocerebellar angle lipoma and the temporal arachnoid cyst; a subtle hemispheric subdural collection, without mass effect, is appreciable on the right.
A CT scan at 4 months of age revealed an increase in size of the right ventricle and corticopial calcifications, and a ventriculoperitoneal shunt was implanted. Follow-up MR studies at 7 and 19 months of age performed with the 3D magnetization-prepared rapid acquisition gradient-echo (MP-RAGE) technique showed a thickened cortex on the right side with few and irregular sulci (Fig 1C), a diffusely hypotrophic corpus callosum, and, in the 19-month study, a subtle subdural collection on the right side (Fig 1D). The other findings were unchanged. A CT study performed at age 3 revealed a further increase in the corticopial calcifications and a small calcification in the right ocular globe of unknown origin; no visual changes were observed. The MR study obtained at age 4 showed a slight enlargement of the right lateral ventricle, prompting surgical revision of the ventriculoperitoneal shunt. The last MR study, performed at age 7, was done with contrast medium and showed subarachnoid-pial enhancement of the right hemisphere, thought to represent leptomeningeal angiomatosis.
Case 2
A male infant of healthy parents was born at term after an uncomplicated delivery. At birth, a scleral mass was noted in the right ocular globe, and cutaneous lesions were present in the right parietal region. A skin biopsy revealed fibro-fatty tissue. Findings at neurologic examination were normal.
A contrast-enhanced CT study, performed at 17 days of life, showed, on the right side, slight enlargement of the lateral ventricle, mainly in the occipital horn, and of the subarachnoid spaces and sylvian fissure; a few corticopial calcifications in the temporooccipital region (Fig 2A); a decrease in size of the cerebral peduncle; and a small arachnoid cyst in front of the temporal lobe. Cranial sonographic and MR studies, obtained at 3 months of age, confirmed the CT pattern. In addition, an MR examination showed a thick cortex with irregular multiple and small gyri in the right temporoparietooccipital region (Fig 2B). This finding was confirmed by a subsequent study performed with the MP-RAGE technique at 16 months of age (Fig 2C). In this last examination, a further widening of the right subarachnoid spaces, a slight reduction in the right lateral ventricle, and a thinning of the corpus callosum were noted. Myelination appeared normal (Fig 2D). A right occipitoparietal chronic subdural hematoma, probably due to a previous head trauma, was also seen; the other findings were unchanged. A CT study, performed at age 2, showed a clear increase in corticopial calcifications, particularly in the temporoparietooccipital region.

Case 2.
A, Contrast-enhanced CT scan obtained at 17 days of life shows a widening of the right lateral ventricle and sylvian fissure and areas of slight hyperdensity in the right temporooccipital region. (Courtesy of Dr. Nadia Colombo, Milan, Italy.)
B, Axial T2-weighted MR image (3000/120/1) obtained at 4 months of age shows an increase in size of the right lateral ventricle and a widening of the right hemispheric subarachnoid spaces. A lack of insular opercularization and cortical dysplasia in the right temporoparietooccipital region is well seen; the abnormal cortex is thicker than normal, with multiple and small gyri.
C and D, MR study obtained at 16 months of age. Coronal reconstruction image obtained with the MP-RAGE technique (C) confirms cortical dysplasia; the temporal cortex appears thick, with irregular gyri. On axial T2 weighted image (3000/120/1) (D), myelination seems to be normal.
Discussion
ECCL is a rare, congenital neurocutaneous syndrome, not inherited in a mendelian fashion. The tissues and organs primarily affected are of ectoderm and mesoderm origin: skin, eye, adipose tissue, and brain. ECCL is limited to one side of the cranium, the face, and brain. The most typical lesions, which occur almost exclusively in this syndrome, are subcutaneous soft tumors, consistent with lipomas, and areas of alopecia; other common findings are ocular lesions, such as defects of the eyelids and epibulbar dermoids (2–8). All these lesions seem to be nonprogressive. Multiple brain malformations on the same side as the head lesions are associated. Usually, seizures develop, beginning in infancy, and affected patients have variable degrees of psychomotor delay and motor impairment (1, 3, 6–10).
The clinical features of ECCL overlap with other neurocutaneous syndromes; the main differential diagnosis includes sebaceous nevus syndrome, oculocerebrocutaneous syndrome (OCC), and Proteus syndrome (3, 4, 6–8). Sebaceous nevus syndrome is associated with linear sebaceous nevi limited to one side of the face, ocular abnormalities, and clinical symptoms in common with ECCL. The most frequent CNS abnormality is widening of the ipsilateral ventricle (11). Sebaceous nevus syndrome and ECCL may thus be a continuum of phenotypic expression (7, 8). OCC syndrome and ECCL share, as well, many clinical features. In OCC syndrome, the presence of orbital cyst, microphthalmia, and skin defects, as well as the absence of facial lipomas and scalp alopecia, are helpful for the diagnosis. In addition, CNS malformations, rare in OCC syndrome, are limited to intracranial cysts and agenesis of the corpus callosum (4). Proteus syndrome is an overgrowth syndrome in which every feature of ECCL can be found; it is bilateral, asymmetric, and involves the head, trunk, and limbs (9, 12). Brain changes are rare (6). In contrast, ECCL is not progressive, unilateral, or limited to the head. Because subcutaneous lipomas below the head and neck, visceral involvement, and hyperostosis of the skull are found in ECCL, some authors suggest that these two syndromes may represent a continuum, and that ECCL may be a localized form of Proteus syndrome (5, 12).
Only a few cases of ECCL have been described since 1970, and little information is available concerning brain anomalies. Only two cases have been reported in which complete neuroradiologic evaluation has been done, and no report has included complete neuroradiologic follow-up.
Our patients were studied with sonography, CT, and MR imaging, and they were followed up for a period of several years. In both children, we found, on the affected side, enlargement of the lateral ventricle, mainly at the occipital horn; widening of the subarachnoid spaces; an arachnoid cyst of the middle cranial fossa; a lack of normal insular opercularization; a dysplastic cortex in the temporoparietooccipital region; corticopial calcifications; and thinning of the corpus callosum. In the first case, we observed a cerebellopontine angle lipoma, a unilateral small ocular calcification, and leptomeningeal angiomatosis. The contralateral hemisphere was normal. All these findings have been variously reported in the literature. In particular, the enlargement of the lateral ventricle, the intracranial cysts, and the atrophy of the cerebral hemisphere are the most constant findings. Some authors have reported the presence of porencephalic cysts as a typical feature of ECCL (3, 6, 7); nevertheless, a careful review of the literature suggests a different interpretation. The areas of porencephaly described were in the temporal or parietooccipital regions and were associated with a dilatation of the ipsilateral ventricle; therefore, these lesions could have been arachnoid cysts of the middle cranial fossa, which do not necessarily have mass effect because they are often associated with a primary hypogenesis of the temporal lobe or are part of a large dilatation of the lateral ventricle. Cortical calcifications and areas of cortical dysplasia have been described as well (1, 3, 4, 7, 9, 12, 13).
The precise neuropathology of cortical dysgenesis cannot be based on neuroimaging findings; the histologic examination performed in one case revealed a polymicrogyric convolutional pattern (1). Intracranial lipomas were found in very few cases (1, 9); in one, a leptomeningeal lipoangiomatosis was also observed (1). A dysmorphic appearance of the corpus callosum (9, 13) as well as unilateral ocular calcifications were found in two cases (3, 4). Our follow-up studies revealed that the hydrocephalus and corticopial and ocular calcifications undergo some change over time. In our first case, there was an increase in ventricular size during the first months of life; in the second case, we observed an enlargement of the lateral ventricle over a period of about 3 months, with a spontaneous, slight reduction in the last study. Other authors (4, 7) have reported cases in which follow-up CT studies performed within the first months of life showed a progressive hydrocephalus requiring a ventriculoperitoneal shunt.
Like the clinical features, the radiologic findings of ECCL also overlap with other syndromes. For example, unilateral cortical calcifications, hemiatrophy, and, sometimes, areas of cortical dysplasia are present in Sturge-Weber disease (SWD). The topographic distribution of the angiomatosis in SWD compares with the extension of the lipomatosis in ECCL. Nevertheless, in SWD the mesodermal dysgenesis is limited to the blood vessels, whereas in ECCL the fat tissue is primarily involved (1). In ECCL, cerebral calcifications are localized throughout the cortex and detectable in the first months of life, whereas in SWD, cortical calcifications are mainly in the occipitoparietal areas and they are seldom evident before 1 year of age. Leptomeningeal angiomatosis (ie, a typical finding of SWD), seen on contrast-enhanced studies in our case 1, has previously been reported in ECCL only in a case in which histologic examination was done (1). In the other reported cases of ECCL, this finding was not described, probably because the absence of contrast enhancement makes visualization of angiomatosis very difficult.
Another differential diagnosis is with unilateral macrencephaly. This malformation is characterized by an overgrowth of one hemisphere and omolateral ventricle, associated with neural migration disorder; in ECCL, the affected hemisphere is clearly atrophic and only the lateral ventricle appears enlarged. Cortical calcifications are rarely detected in unilateral macrencephaly. In addition, neither intracranial lipomas nor arachnoid cysts occur in SWD or in unilateral macrencephaly. Both our patients had normal neurologic findings at birth; the first child developed slight left hemiparesis, mild mental retardation, and epilepsy with time. Even though clinical features of ECCL have variable degrees of severity, only a few cases have been reported in which there is normal mental development and no seizures (3, 5, 8).
Conclusion
ECCL syndrome has a quite specific neuroimaging pattern that is well seen with MR imaging, CT, and sonography; the use of contrast medium is suggested.
Footnotes
↵1 Address reprint requests to C. Parazzini, MD, Department of Neuroradiology, Scientific Institute H. S. Raffaele, University of Milan, Via Olgettina 60, 20132 Milan, Italy.
References
- Received August 28, 1997.
- Accepted after revision May 19, 1998.
- Copyright © American Society of Neuroradiology








{kind=link}
{kind=link}