
Abstract
BACKGROUND AND PURPOSE: The etiology of the neurotoxicity associated with cyclosporin-A (CsA) and FK-506 treatment is not fully understood. At our institution, we noticed a distinct, abrupt change in the imaging characteristics of CsA and FK-506 neurotoxicity, which consisted of a shift in lesion morphology from a white matter abnormality to a mixed cortical and white matter pattern. The purpose of this study was to assess clinical parameters that might explain this change.
METHODS: Twenty-two patients had a neurotoxic reaction and brain imaging changes while receiving CsA or FK-506. Nineteen patients received allogeneic bone marrow transplants, and three had aplastic marrow disorders. Fifty-one imaging studies (CT or MR imaging) were obtained, and lesion characteristics, locations, and time courses were evaluated along with relevant clinical data.
RESULTS: Nine patients who had been conditioned for transplantation with cyclophosphamide and chemotherapy (busulfan or thiotepa) had a mixed pattern of cortical and white matter involvement (57 lesions). Isolated white matter involvement (62 lesions) developed in three nontransplant patients and 10 transplant patients conditioned with cyclophosphamide and total-body irradiation. All lesions occurred at typical brain watershed zones. Lesion enhancement was noted in two patients conditioned with chemotherapy. Initial images demonstrated characteristic lesions in 15 patients (68%). Initial images were normal in four patients (18%) and nonspecific in three patients (14%).
CONCLUSION: Lesion location in CsA and FK-506 neurotoxicity may depend on the presence or type of conditioning used before bone marrow transplantation. Nontransplant patients or those conditioned with total-body irradiation develop white matter lesions, whereas those conditioned with chemotherapy develop mixed cortical and white matter lesions.
Cyclosporin A (CsA) and FK-506 are immunosuppressive agents used to control transplant rejection and graft-versus-host disease (GVHD). Reports have linked these two drugs with central nervous system toxicity, which clinically varies from tremulousness to grand mal seizure activity (1–33).
Subcortical and deep white matter changes on imaging studies have been the alterations most commonly described (17, 18, 21–23, 26–31). Cortical involvement has been noted, and contrast enhancement occasionally seen (23, 27, 30, 32, 33). This pattern occasionally has been referred to as the “reversible posterior leukoencephalopathy syndrome” (34, 35). Nonspecific white matter features have also been described (36).
The etiology of CsA and FK-506 neurotoxicity is not understood. No single factor has been consistently identified in all patients. Direct toxicity has been postulated, but blood CsA levels usually are within the therapeutic range in most patients (2–4, 6, 8, 9, 15, 16, 21, 23–25, 27, 29–32). Abnormal CsA metabolism due to hepatic dysfunction or hypocholesterolemia, leading to altered CsA binding and secondary increase in brain drug distribution, also has been considered (18, 24). Some have postulated that endothelial damage plays a role, with the release of vasoactive peptides leading to labile blood pressure and vasospasm (28, 32, 33, 37, 38), in addition to thrombotic microangiopathy leading to microvascular damage (31, 39). Hypertension has been suggested as a primary cause of the brain changes and has been associated with increased sympathetic neural activation (30, 40, 41). Selected reports have considered other potential causes, such as high-dose methylprednisolone therapy (2, 4), ketoconazole therapy (3), hypomagnesemia (9), anaphylactic reactions (8), and human leukocyte antigen (HLA) mismatch (33).
We recently observed a change in the imaging appearance of neurotoxicity in patients receiving an allogeneic bone marrow transplant (BMT) at our institution. This change was associated with a change in the treatment protocol. Previously, we typically observed white matter changes in our patients with CsA or FK-506 neurotoxic reactions. When the pretransplantation conditioning protocols used in the BMT service at our institution were changed, we began to observe the presence of cortical involvement. The purpose of this study was to review the imaging and clinical features of 22 patients with CsA or FK-506 neurotoxic reactions in an effort to determine factors that underlie cortical or white matter lesions.
Methods
One hundred eighty-six patients received an allogeneic BMT at our institution during 6 years. Patients were receiving CsA or FK-506 to prevent GVHD. In 19 of these patients, notable neurologic symptoms developed, and imaging studies showed brain changes consistent with previous descriptions of CsA or FK-506 neurotoxicity. Three additional nontransplant patients receiving CsA for aplastic marrow disorders also developed neurotoxic reactions. These 22 patients form the subject of this report.
Ten patients were female and 12 were male, with an average age of 45 y (range, 19 to 65 y). Thirteen of the allogeneic BMT patients received transplants from related donors, and six received transplants from matched unrelated donors. Nontransplant patients 20 and 21 received CsA for treatment of severe aplastic anemia, and patient 22, for red blood cell aplasia. Solid-organ transplantation is not performed at our institution. A summary of the reasons for transplantation in these patients is presented in Table 1.
Reason for BMT or CsA and FK-506 treatment
BMT Procedures
Patients underwent BMT in accordance with treatment protocols approved by the internal review board at our institution. CsA or FK-506 and methylprednisolone (1.0 mg/kg/d tapered to 0.1 mg/kg/d) were used as prophylaxis against GVHD. CsA doses were adjusted to maintain whole-blood CsA levels of 350–600 μg/L as measured with polyclonal fluorescence polarization assay. Patient 19 received 0.03 mg/kg FK-506. Patients 10, 13, and 14 initially received CsA, but this was changed to FK-506 when CsA-related toxicity developed.
Preparative Regimens
Conditioning eliminates the host's bone marrow in preparation for receiving the donor's marrow. Three conditioning regimens were used for the BMT patients. Ten patients received conditioning with cyclophosphamide (50–60 mg/kg/d for 2–4 d), followed by total-body irradiation (TBI) (300 cGy/d for 4 d). Four patients received conditioning with cyclophosphamide (60 mg/kg/d) and thiotepa (5 mg/kg/d), followed by TBI (300 cGy/d) for 4 d. Five patients received oral busulfan (4 mg/kg/d for 4 d) and cyclophosphamide (60 mg/kg/d for 2 d).
Imaging Procedures
Fifty-one imaging studies were available for evaluation: 33 CT scans and 18 MR images. Contrast-enhanced studies were obtained in 24 CT and 11 MR imaging examinations. CT scans were obtained with 5-mm contiguous images obtained through the posterior fossa and 10-mm images obtained to the vertex. Contrast material, when used, consisted of a 150-mL bolus infused through peripheral venous access. MR images were obtained with a 1.5-T imager. Sagittal T1-weighted scout views were followed by axial T1-weighted images (600/25/1 [TR/TE/excitation]) with 5-mm section thickness and axial proton density–weighted and T2-weighted images (2500/25–84/2) with 5-mm section thickness. Contrast-enhanced T1-weighted images were obtained by using 0.1 mmol/kg gadopentetate dimeglumine and typical T1-weighted parameters, as described above. When fast spin-echo images became available, sequences were performed with similar TR, TE, and section-thickness parameters. The echo train length on T1-weighted and proton density–weighted images was four; the echo train length on T2-weighted images was eight.
In one patient, 3D time-of-flight (TOF) MR angiograms were obtained (46/6.9/1) with 0.9-mm section thickness, an 18-cm field of view, and a 30° flip angle before and after contrast enhancement to assess the intracranial vessels.
Three neuroradiologists (W.S.B., M.P.S., L.L.) retrospectively reviewed the CT and MR images. The neuroradiologists were aware that these were images obtained from BMT patients or those receiving CsA or FK-506 treatment, but they were blinded to the use and type of conditioning regimen, if any. The brain lesions were identified, and their location and characteristics were decided on by consensus. The images depicted lesions that were separate in location or that began as separate and became confluent over time. At CT, lesions had low attenuation. At MR imaging, lesions typically had high signal intensity on proton density–weighted and T2-weighted images and low signal intensity on T1-weighted images.
Lesion location within the brain parenchyma (cortex vs white matter) was assessed, and white matter abnormality versus a mixed cortex and white matter lesion location was determined by means of visual assessment of lesion location on contiguous sections. The objective was to determine if the abnormality appeared in only white matter, only cortex, or both in a given region. The assessment of subtle variations in lesion position at the junction of cortex and white matter was considered beyond the capabilities of available techniques.
At CT, a lesion was labeled as exclusively in white matter if the white matter had low attenuation and if the overlying cortex could be identified as being separate from the underlying white matter abnormality. Edema often would stretch or distort the overlying cortex, but cortical attenuation was judged normal. A lesion was labeled as exclusively in the cortex if abnormally low attenuation was present at the brain periphery along the cortical surface without explicit underlying white matter abnormality. A lesion was labeled as mixed cortex and white matter if the white matter was abnormal in attenuation and the abnormality was seen to extend clearly to the cortical surface on several contiguous sections.
At MR imaging, lesion location was judged by assessment of contiguous sections with the added value of signal intensity assessment and location clarity rendered with available MR sequences. The location and limits of the cortex typically are visible on MR images, particularly on T2-weighted images. Axial T1-weighted, proton density–weighted, and T2-weighted images were interpreted together for the best identification of the lesion position, particularly the position relative to the extracerebral CSF space. Therefore, lesion location was judged considering the position relative to visualized cortex, the position relative to the underlying subcortical white matter, the position relative to the brain surface and extracerebral CSF, and the appearance on multiple contiguous axial sections.
On MR images, lesions located exclusively within the white matter had high signal intensity on T2-weighted images, with various quantities of edema. The adjacent cortical surface might have been deformed or compressed by underlying edema, but this change could be clearly identified. The cortical signal intensity was judged normal compared with that of other regions of uninvolved cortex available for comparison. Lesions were considered to be exclusively within the cortex if the cortex signal intensity was judged abnormal and if no abnormality could be identified in the adjacent white matter. Lesions were considered to involve both the cortex and white matter if the signal intensity abnormality coursed through the white matter and extended into the cortical surface.
Supratentorial lesions were consistently identified in four primary locations: frontoparietal junction, parietal region, occipital poles, and inferior temporo-occipital junction. Lesions, therefore, were tabulated in these common regions. Additional lesions were identified less often in the cerebellar hemispheres, splenium of the corpus callosum, corona radiata, and frontal lobes. We occasionally identified small, nonspecific, focal white matter changes. These lesions appeared randomly located, and no attempt was made to itemize these areas.
Clinical Review
Inpatient and outpatient records of these 22 patients were retrospectively reviewed. Factors assessed with regard to CsA and FK-506 toxicity included elevated blood pressure, CsA level, magnesium level, cholesterol level, HLA matching, GVHD, veno-occlusive disease, and BMT-related thrombotic microangiopathy (TM). The presence of seizure activity was noted, including timing of neurotoxic reaction relative to brain imaging. Baseline blood pressure and blood pressure at the time of toxic reaction were recorded.
The presence of BMT-related TM, GVHD, and veno-occlusive disease of the liver were noted and graded by using techniques previously described (42–45). Blood vessel endothelial injury is suggested clinically when patients receiving an allogeneic BMT have evidence of transplant-related TM (42). Clinical BMT-related TM (grades 2–4) was diagnosed and graded if the lactate dehydrogenase (LDH) level was increased in association with 1.3–4.8% schistocytes for grade 2, 4.9–9.6% schistocytes for grade 3, and schistocytes of 9.7% or more for grade 4, as previously described (42).
For the determination of fragmented erythrocytes (schistocytes), a single observer counted 500 red blood cells on smears at the time of patient evaluation. The proportion of fragmented red blood cells then was calculated. Acute GVHD was diagnosed and staged from I to IV according to the Seattle criteria (43, 44). Staging was confirmed histologically with skin, gut, or liver biopsy. Tissue evidence of vascular inflammation is a component of the grading system of acute GVHD.
As proposed by Jones et al (45), veno-occlusive disease of the liver was diagnosed if the bilirubin level was 2 mg/dL or higher, with two of the following: hepatomegaly, ascites, or weight gain of 5% or more. Veno-occlusive disease was graded as mild (resolved without therapy), moderate (resolved with treatment), or severe (unresolved or the patient died before day 100 after BMT).
Results
Conditioning regimens and relevant laboratory data are presented in Table 2. Imaging changes typical of CsA and FK-506 neurotoxicity developed in 19 (10.2%) of 186 allogeneic BMT patients (13 of 133 with related donor transplants and six of 53 with matched, unrelated donor transplants). The typical changes of CsA and FK-506 neurotoxicity included patchy or confluent areas of white matter, cortical, or mixed abnormality with or without edema in the occipital lobes, parietal region, frontoparietal junction, and inferior temporo-occipital junction. In the three nontransplant patients, a clinical neurotoxic reaction and brain imaging changes were similar to those of the transplant group.
Clinical data
Clinical Results
The average duration of CsA and FK-506 treatment at the time of presentation was 70 d (range, 2–480 d). In 15 patients, symptoms occurred within the first month of treatment, whereas in two patients, a toxic reaction developed 12 and 16 mo after transplantation. Seizure was the presenting problem in 16 patients; cortical blindness, in one; and confusion, agitation, or headache in the remaining five.
Sixteen of 19 allogeneic BMT patients were evaluated for BMT-related TM. All of these had positive test results for abnormal red blood cell morphology. Thirteen of 16 patients had severe (grade 3–4) BMT-related TM, and the other three patients developed moderate (grade 2) BMT-related TM. The nontransplant patients were not evaluated for microangiopathic hemolytic anemia. Four BMT patients had veno-occlusive disease. Ten of 15 BMT patients evaluated had acute GVHD (grades 3–4), and another patient had evidence of chronic GVHD. Three patients did not survive long enough for acute GVHD to develop.
HLA compatibility is listed in Table 2. Thirteen patients received marrow from matched (5/6 or 6/6 antigenic loci) related donors, one patient received marrow from a weakly matched (4/6) related donor, four patients received marrow from matched (5/6 or 6/6) unrelated donors, and one patient received marrow from a weakly matched (4/6) unrelated donor. Seventeen of the 19 BMT patients received marrow from matched (5/5 or 6/6) donors.
Most patients had mildly to moderately elevated blood pressure at the time of toxic reaction (Table 2). Renal function was mildly abnormal in most patients; the creatinine level was higher than 2.0 mg/dL in four patients. The magnesium level was elevated in one patient, and CsA levels were within the therapeutic range in all but three patients (patients 1, 13, and 16) at presentation. Two patients were receiving FK-506 at the time of toxic reaction. In one, the FK-506 level was 59.5 ng/mL (normal range, 5–20 ng/mL) 3 d before toxic reaction, but the level was normal at the time of imaging. In the other, the FK-506 level was not obtained at the time of toxic reaction. Cholesterol levels were normal or elevated in all patients. Eight patients were receiving high-dose steroids at presentation.
Imaging Results
The morphologic features and distribution of lesions in CsA and FK-506 toxicity are summarized in Tables 3 and 4. One hundred nineteen lesions were identified in the 22 patients. Lesion locations conformed to a watershed distribution. The most common sites were in the occipital pole and parietal regions and less often in the frontoparietal junction and inferior temporo-occipital junction. In three patients, five cerebellar lesions were identified. In one patient, the insular region was involved with extension into the corona radiata. Three patients had unusual lesions in the splenium of the corpus callosum, likely the junction between the pericallosal artery arising as a branch of the anterior cerebral artery and the posterior pericallosal artery, a branch of the posterior cerebral artery (Fig 1B). Two distinct types of lesions were noted (Table 4) and labeled group 1 and group 2 for ease of discussion. Group 1 lesions showed subcortical and deep white matter abnormalities without involvement of the adjacent cortex. Group 2 lesions showed a mixed pattern with primarily cortical involvement with various involvement of the subcortex and deep white matter.
Lesion distribution and morphology
Summary of lesion distribution in groups 1 and 2

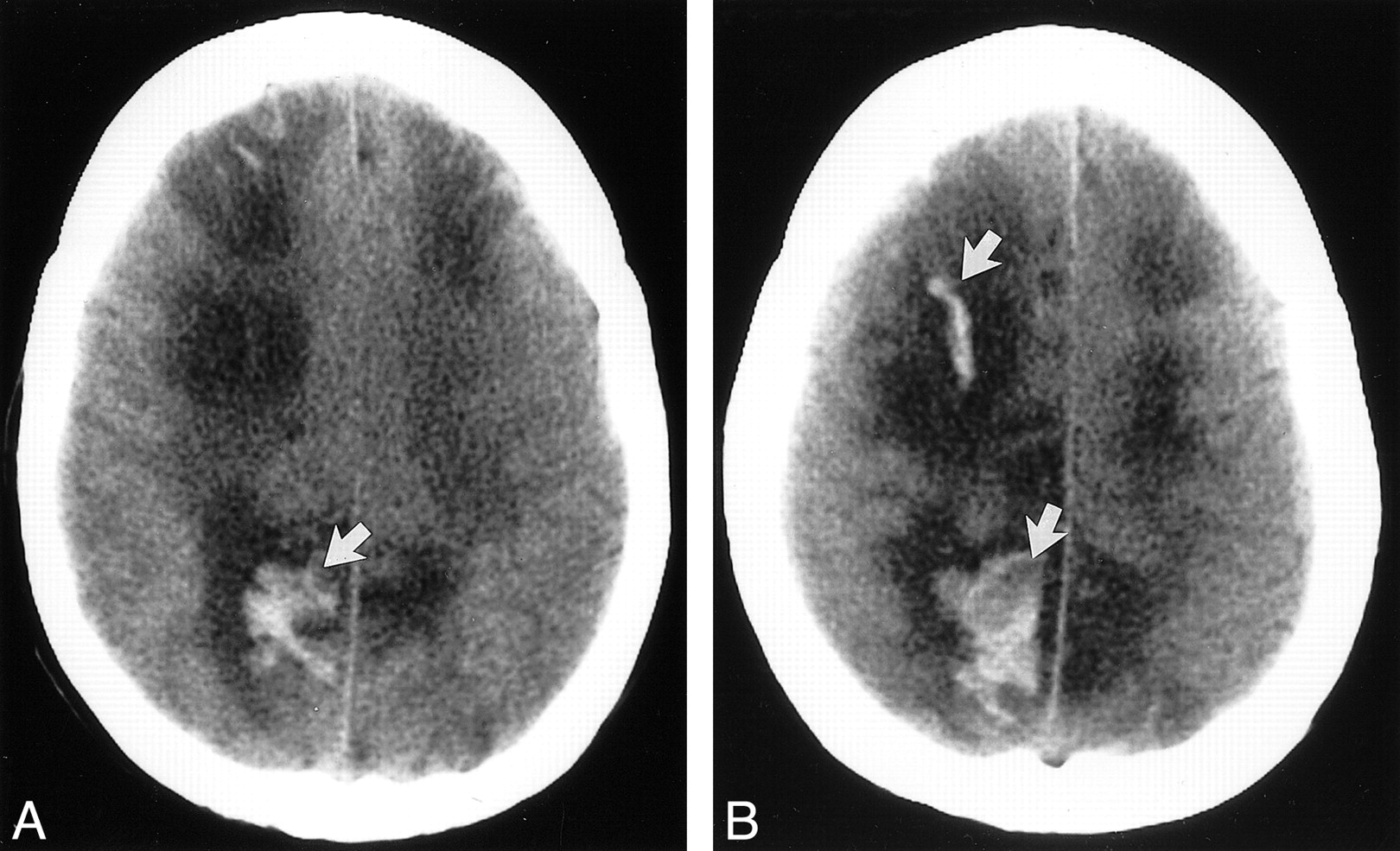
fig. 1. Axial nonenhanced CT sections obtained 24 h after seizure in a patient (case 4) who was conditioned for transplantation with cyclophosphamide and TBI. She had a new-onset seizure 23 d after transplantation. At the onset of toxic reaction, she had grade 3 BMT-related TM, and her CsA level was within the normal therapeutic range. The initial head CT study obtained on the day of the seizure was normal. Her baseline blood pressure was 92/52 mm Hg but increased to 230/130 mm Hg at the time of toxic reaction and initial CT.
A, Image at the level of lateral ventricles shows edema in the corpus callosum (arrow).
B, Image at the level of the sentrum semiovale shows extensive areas of white matter edema in the parietal region bilaterally (arrow).
Group 1 Subcortical White Matter Lesions
In three nontransplant patients and 10 BMT patients conditioned with cyclophosphamide and TBI, CT or MR images showed typical subcortical white matter abnormality, as previously described (17, 18, 21–23, 26–31). Most patients had numerous and bilateral lesions. The lesions had low attenuation on CT scans. This subcortical white matter abnormality often had strikingly low attenuation, which resembled the changes seen in tumor edema. On MR images, the subcortical white matter lesions were high in signal intensity on proton density–weighted or T2-weighted images (Fig 2). These lesions were slightly low in signal intensity on the T1-weighted images, rendering poor definition of the gray matter–white matter junction. Neither CT nor MR images showed contrast enhancement in any lesion in these patients. This group had no cortical involvement, but abnormal signal intensity did extend to the gray matter–white matter interface.

fig. 2. T2-weighted MR image (2500/84/2) obtained on the day of presentation shows typical areas of subcortical white matter abnormality without cortical involvement (arrows) in patient (case 2) who was conditioned for transplantation with cyclophosphamide and TBI and who presented with confusion and altered mental status 14 d after transplantation. She did not develop seizures. At the time of toxic reaction, she had grade 4 BMT-related TM and a CsA level within the normal therapeutic range. Her baseline blood pressure of 115/85 mm Hg increased to 167/104 mm Hg at the time of toxic reaction and MR imaging
Many lesions showed edema, with distortion and compression of the overlying cortex. The imaging appearance of the lesions depended on the severity of edema. Two patients had severe edema with very thin but clearly defined cortex. Another two patients had moderate edema with thinning of the adjacent cortex. The remaining nine patients had mild white matter edema with well-defined cortex adjacent to the subcortical white matter abnormality. As the subcortical white matter edema increased, less of the cortex could be seen. With severe edema, only a thin ribbon of cortex could be seen. The cortex often appeared thinned or compressed, but cortical abnormalities seen in brain infarction were not present.
Acute hemorrhage was identified in a single patient with group 1 lesions. This patient presented with only a headache. The baseline blood pressure was 140/82 mm Hg, and at the time of clinical presentation was moderately elevated to 164/106 mm Hg. In a second patient in this group, MR images depicted a subtle region of nonspecific older hemorrhage in the posterior fossa.
Group 2 Mixed Cortical and Subcortical White Matter Lesions
Nine BMT patients had mixed lesion morphology, with primarily cortex abnormality and various subcortical and deep white matter involvement (Fig 3A–C). This pattern was identified exclusively in the patients conditioned with cyclophosphamide and busulfan or cyclophosphamide, thiotepa, and TBI. On MR images, the cortex signal intensity was abnormal on the proton density–weighted and T2-weighted images. Slight cortical or white matter hypointensity also was noted on the T1-weighted images in these patients. Subcortical white matter abnormality was identified adjacent to or in addition to the cortical lesions, but to a far lesser extent than in the group 1 patients. Some local cortical edema was noted in these lesions but substantial mass effect was not.

fig. 3. MR images in a patient (case 14), conditioned for transplantation with cyclophosphamide and busulfan, who presented with new-onset seizure 176 d after transplantation. The patient had grade 3 BMT-related TM that rapidly progressed to grade 4. The CsA level was in the normal therapeutic range. His baseline blood pressure was 160/90 mm Hg, which increased to 170/110 mm Hg at the time of toxic reaction. The initial CT scan showed only nonspecific areas of low attenuation in the parietal region. MR images were obtained 6 h after CT.
A–C, T2-weighted MR images from the midventricular level and high parietal region (2500/84/2) show extensive high-signal-intensity edema involving the cortex in the frontal and parietal regions bilaterally (arrowheads).
D and E, Contrast-enhanced images corresponding to B and C show bilateral punctate enhancement in the cortex of the frontal region (arrows). These areas of enhancement correspond to the regions of cortical signal abnormality noted in B and C.
Focal areas of hemorrhage developed in one patient in this group but contributed only slightly to the local mass effect observed (Fig 4). This patient presented with seizure. The baseline blood pressure was 110/70 mm Hg, and that at presentation was 152/96 mm Hg. No notable hematoma evolved.

fig. 4. CT scans obtained at the time of toxic reaction shows areas of low attenuation extending centrally from the frontal lobe to the parietal region, involving the white matter and projecting to the cortical surface. Hemorrhages are noted in the right frontal lobe and right parietal area (arrows). The brain abnormalities are severe and appear to lie in a region that could easily coincide with the boundary between anterior cerebral and middle cerebral territories. Images were obtained in a patient (case 11), conditioned for transplantation with cyclophosphamide and busulfan, who presented with generalized tonic-clonic seizure activity 8 d after transplantation. At the onset of toxic reaction, the patient had grade 3 BMT-related TM that rapidly worsened to grade 4. The cyclosporin level at the time of toxic reaction was within the normal therapeutic range. Her baseline blood pressure was 110/70 mm Hg, which increased to 152/96 mm Hg at toxic reaction
In three patients with group 2 lesions, MR images showed cortex enhancement with a fine stipple pattern present along the brain surface (Fig 3D and E). Seven lesions were present in one patient receiving CsA and conditioned with cyclophosphamide and busulfan, including one lesion in the left cerebellar hemisphere. Three enhancing lesions were present in a second patient receiving CsA and conditioned with cyclophosphamide, thiotepa, and TBI—two in the cerebellum and one in the inferior temporo-occipital junction. Bilateral enhancing frontal lesions were present in a third patient receiving FK-506 and conditioned with cyclophosphamide, thiotepa, and TBI. The pattern of enhancement was always superficial, never bandlike or gyral, as typically is seen with cerebral infarction. Contrast-enhanced CT scans were not obtained at the time of MR imaging in these three patients.
Lesion Evolution
Lesions characteristic of CsA and FK-506 toxicity were present on the initial imaging examination (11 CT, four MR imaging) in 15 patients. In four patients, the initial CT scan was negative, and the lesions were identified on follow-up CT scans. In one of these four patients, the initial CT scan was negative, but follow-up CT scan obtained 24 h later clearly depicted extensive subcortical white matter changes (Fig 1A and B). In three patients, the initial scan showed nonspecific findings that became typical for CsA toxicity on follow-up scans. In one of these patients, the initial MR image showed nonspecific white matter changes that became typical for CsA toxicity on follow-up CT scans. In two patients, late follow-up images showed complete or near-complete lesion resolution.
MR Angiography
One patient underwent MR angiography (Fig 5). MR images in this patient showed a typical group 2 pattern, with cortical and subcortical white matter lesions in the parietal region on the left and occipital pole bilaterally. The MR angiogram showed reduced caliber of the bilateral middle and posterior cerebral arteries, with an appearance like a string of beads in the posterior cerebral artery on the left. Also, visualization of the posterior branch of the cerebral artery may have been reduced. This arterial pattern suggested vasospasm or a vasculitic involvement.
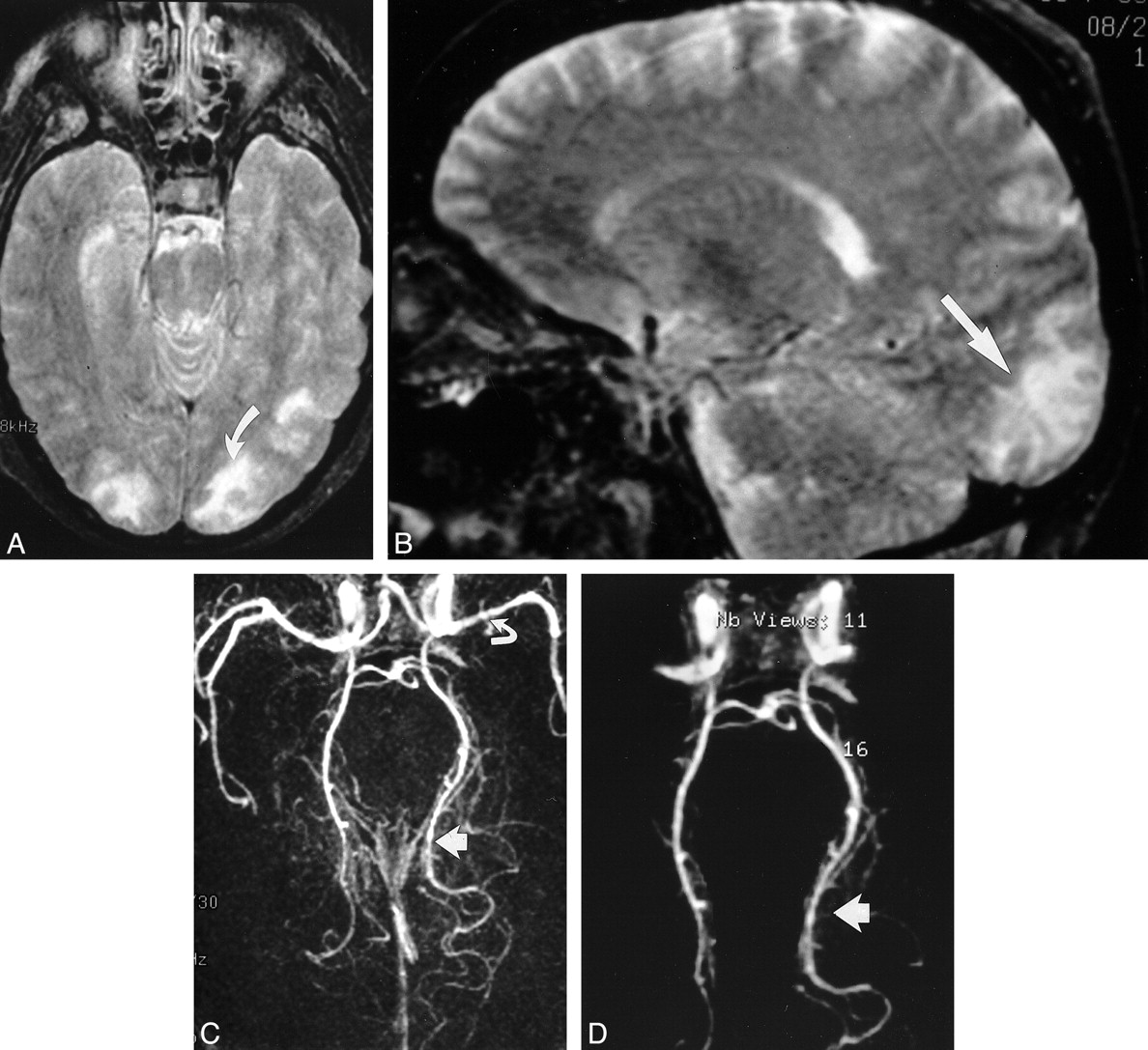
fig. 5. Images in a patient (case 18), who was conditioned for transplantation with cyclophosphamide, thiotepa, and busulfan and who presented with binocular cortical blindness 15 d after transplantation. At the time of toxic reaction, the patient had grade 3 BMT-related TM, and the CsA level was within the normal therapeutic range. Her baseline blood pressure was 150/88 mm Hg, which increased to 180/100 mm Hg at toxic reaction.
A, T2-weighted axial MR image (2500/84/2) shows areas of mixed cortical and white matter signal intensity abnormalities in the occipital pole region bilaterally. Abnormalities are greater on the left (arrow). At autopsy, vasogenic edema was present in the bilateral occipital lobes, with evidence of ischemic neuronal necrosis in the cortex at the occipital watershed region.
B, T2-weighted sagittal MR image (2500/84/2) clearly shows signal intensity abnormality in the calcarine region (arrow).
C, 3D TOF MR angiogram, collapsed view (46/6.9/1), shows vessel narrowing and irregularity in the left middle cerebral artery (curved arrow) and bilateral posterior cerebral arteries; these findings are more striking on the left (straight arrow).
D, Postprocessed 3D TOF MR angiogram with background noise elimination shows a string-of-beads appearance in the posterior cerebral arteries; the finding is more severe on the left (arrow). At autopsy, these vessels appeared normal, without evidence of vasculitis.
Pathology
Findings from brain pathologic evaluation at autopsy were available for three patients. In patient 18, who was conditioned for transplantation with cyclophosphamide, busulfan, thiotepa and TBI, brain pathologic findings obtained 5 d after imaging showed deep white matter and subcortical white matter vasogenic edema, which was most prominent in the occipital poles bilaterally. Areas of cortical ischemia, neuronal necrosis, and petechial hemorrhage were noted and were most prominent in the arterial border zones and at the depths of the sulci. Vasculitis or white matter demyelination was not noted. In patient 8, who was conditioned with cyclophosphamide and TBI, the brain appeared normal 6 mo after imaging. In patient 2, who was conditioned with cyclophosphamide and TBI, brain pathologic findings obtained 1 mo after imaging showed the focal area of older hemorrhage in the posterior fossa depicted on MR images, but they were otherwise normal.
Discussion
The imaging characteristics in our patients with CsA and FK-506 neurotoxic reaction depended on the conditioning regimen used before allogeneic BMT. Patients conditioned with cyclophosphamide and a chemotherapeutic agent (busulfan or thiotepa) had cortical abnormalities with various degrees of subcortical and deep white matter involvement. Nontransplant patients and transplant patients conditioned with cyclophosphamide and TBI had lesions in the deep and subcortical white matter, without apparent cortical involvement. This observation suggests that pretransplantation conditioning regimens may play a role in the response of the brain to CsA and FK-506. While this difference may provide a clue to the pathophysiology of CsA and FK-506 neurotoxicity, its cause is not understood and remains controversial.
The process of allogeneic BMT is complex. Patients typically have a malignancy that involves the bone marrow. Conditioning with either TBI or chemotherapy destroys the patient's native bone marrow and is followed by marrow replacement from an immunologically matched donor by means of allogeneic transplantation. Drugs such as CsA or FK-506 are administered in an effort to prevent GVHD. Despite these treatments and close immune matching, GVHD still may occur. Posttransplantation problems include infection control, renal dysfunction, hepatic dysfunction, metabolic problems (such as hypomagnesemia and hypocholesterolemia), blood pressure control, and ongoing control of graft-related complications.
It is unclear whether the clinical and imaging changes seen in CsA and FK-506 neurotoxicity relate to direct drug toxicity, metabolic disturbances (hypomagnesemia or hypocholesterolemia), hypertension, vascular damage, or a combination of these factors. Several findings, such as the presence of schistocytes and the thrombomodulin–creatinine ratio, suggest central vascular damage, which causes leakage of drug and edematous fluid into brain tissue, with hypertension occurring as a secondary phenomenon.
BMT thrombotic microangiopathy is a complication of marrow transplantation characterized by abnormal red blood cell morphology (schistocytes), which indicates endothelial injury (39, 42). BMT-related TM may remain as a mild form, with only an increase in fragmented red blood cells and an elevated LDH level, or it may worsen to include fever, neurologic changes, and renal changes progressing to severe multiorgan failure. All allogeneic BMT patients have evidence of BMT-related TM (42).
Thrombomodulin is a transmembrane receptor for thrombin, and it is released into the blood stream only with endothelial damage (46). Thrombomodulin is excreted by the kidneys; therefore, an elevated thrombomodulin/creatinine ratio is additional indirect evidence of endothelial injury. Thrombomodulin/creatinine ratios are elevated in patients with BMT-related TM; this finding again indicates that endothelial damage is a feature of BMT-related TM (46).
Taken together, the above information suggests that CsA and FK-506 neurotoxicity is a manifestation of a more widespread vasculopathy. CsA and FK-506 can injure the endothelial cell directly (37–39, 47–49). Endothelial cell damage could be responsible for direct injury to the capillary bed, for altering the blood-brain barrier, as well as for the release of potent vasoconstrictors such as endothelin or thromboxane, resulting in vasospasm (28, 32, 33, 37, 38). Several authors have suggested that injury to the blood-brain barrier may occur in CsA toxicity (13, 22, 26). Two recent reports note brain enhancement in several patients (30, 33). Vasoconstriction and vasospasm leading to local ischemia also has been proposed as a potential contributing mechanism to CsA and FK-506 neurotoxicity (28, 32, 38).
The variation in lesion morphology with transplantation conditioning (white matter vs mixed cortical and white matter involvement) may be a clue to the mechanism of CsA and FK-506 toxicity. Chemotherapeutic agents might render the cortical endothelium more susceptible to the toxic effects of CsA and FK-506. Capillary endothelial injury could cause altered microvascular permeability, leading to vasogenic edema in the white matter or the cortex. Subtle cortical enhancement could occur when the process is severe. Cortical enhancement was noted in two of our patients induced with chemotherapy (one induced with cyclophosphamide and busulfan; one induced with cyclophosphamide, thiotepa, and TBI). The unique enhancement pattern shows a stippled appearance (not gyral, as is present in brain infarction) and confirms breakdown of the blood-brain barrier.
If central arterial and diffuse local vasospasm were present, reduced brain blood flow would occur, in particular at the watershed zones, and could explain the minor ischemic changes noted at histologic evaluation (28, 32, 33). The lesions in CsA toxicity predominate in four major regions in the cerebral hemispheres: occipital poles, parietal region, frontoparietal junction, and inferior temporo-occipital junction. This pattern corresponds to the watershed zones between major vascular territories and their main branches (31). The watershed distribution suggests that local blood flow in the brain is altered (reduced) and that a blood vessel or arterial process is rendering the boundary zones between major arterial territories most vulnerable.
Vasospasm alone probably is inadequate to produce the lesions present in CsA and FK-506 toxicity (28). Severe hypoperfusion alone would likely lead to brain infarction (50). Modest hypoperfusion and brain ischemia coupled with a second process seems more plausible in the absence of brain infarction. One cannot rule out a unique drug effect in the face of hypoperfusion. The chemotherapeutic drugs (cyclophosphamide, busulfan, thiotepa) are not known to promote blood vessel abnormalities or vasculitis. Drug toxicity from either chemotherapy alone or from CsA and FK-506, however, may effect the brain differently under conditions of hypoperfusion. Under conditions of hypoperfusion and ischemia induced by vasospasm, drugs such as CsA and FK-506 or chemotherapeutic agents could damage the blood-brain barrier. Whatever the mechanism, brain changes can occur rapidly in these patients, as evidenced in patient 4, whose CT scans were normal on the day of toxic reaction (seizure) but showed extensive areas of white matter edema only 1 d later (Fig 1).
Consequences of endothelial injury might help explain several features of CsA and FK-506 neurotoxicity noted in one of our patients. The abnormal posterior cerebral arteries shown on MR angiograms in patient 18 points to an arterial abnormality. This patient had BMT-related TM (grade 3; 4.9–9.6% schistocytes), and her blood pressure at the time of toxicity was only mildly elevated (180/100 mm Hg). At autopsy, the large and medium arteries in this patient were normal, with no evidence of vasculitis or inflammatory change. In the absence of vessel inflammation, the MR angiographic appearance may represent vasoconstriction or vasospasm secondary to vasoconstrictor release as a result of endothelial injury as has been suggested (28, 32, 33, 38). Similar MR angiographic features have been noted in patients with preeclampsia or eclampsia (51, 52).
Brain pathologic findings of CsA neurotoxicity is limited, but blood vessel inflammation and minor ischemic changes have been identified, along with old hemorrhage and myelin pallor (13, 14, 21, 27). At autopsy performed 5 d after the onset of neurotoxic reaction, brain pathologic findings in patient 18 showed moderate vasogenic edema in the occipital poles, with ischemic neuronal necrosis present in the watershed regions. The ischemic neuronal necrosis likely is evidence of watershed hypoperfusion. These findings parallel the histologic changes encountered secondary to global ischemia (50). Vasogenic edema could be related to microscopic endothelial injury secondary to disruption of the blood-brain barrier. This process may be similar to the vasogenic edema recently identified at diffusion MR imaging in a patient with eclampsia (53).
We saw no lesion enhancement in our group 1 patients. CT images in these patients frequently showed cortical hyperattenuation, but this seemed related to gyral compression caused by underlying subcortical edema. Most lesions showed slight or mild edema with only local mass effect and effacement of local cortical sulci. The edema in CsA and FK-506 toxicity resembles the pattern seen in eclampsia. It may represent vasogenic edema, as has been noted (53). In two patients, moderate lesion edema was present with substantial flattening and thinning of local cortical gyri and local mass effect. This included a single lesion within the cerebellum. In two patients, severe edema resulted in notable ventricular deformity. Ventricular obstruction from brain edema was not seen in our patients but has been reported (29).
In a recent study, hypertension was suggested as the cause of CsA or FK-506 neurotoxicity (30). But hypertension is not a constant finding at the time of neurotoxicity, and almost all patients with BMT-related TM have hypertension, to various degrees. These recent study findings suggest that the brain lesions present in CsA neurotoxicity are related to brain autoregulation being overwhelmed, with local perfusion capillary breakthrough (30). This process was thought to reflect a combination of local reduction in brain sympathetic innervation and hypertension from increased sympathetic activity in patients receiving CsA (30, 40, 41). Unfortunately, blood pressure data in this article were presented as an averaged mean (30), and individual blood pressure measurements at the time of toxic reaction were not reported. In our patient population, hypertension was common at the time of toxic reaction, but it was not present in all patients (Table 2). This finding parallels the data reported in the literature. In the articles in which blood pressure effects are clearly identified, hypertension was noted in 47 (73%) of 64 patients (1, 2, 4, 5, 9, 15, 17, 18, 25, 27, 31). But of equal importance, hypertension was not present in 17 (27%) of these 64 patients, and no change in blood pressure was noted at the time of toxic reaction (4, 9, 20, 23, 24, 28, 31, 33). Absence of hypertension in these patients makes it difficult to accept elevated blood pressure as the single cause of CsA and FK-506 neurotoxicity.
Systemic effects of endothelial injury could be acting on the brain and other organs concurrently. The cause of hypertension could be multifactorial in these patients. Hypertension could be secondary to systemic endothelial or vascular injury (BMT-related TM), with the release of vasoactive substances (such as endothelin or thromboxane) rather than being the singular cause of CsA and FK-506 neurotoxicity (37, 38, 54–57). Hypertension could contribute to the brain changes in CsA and FK-506 neurotoxicity. The involved blood vessels could be functionally abnormal, and adverse effects may be present due to failed or suboptimal attempts to autoregulate. Blood vessel and capillary endothelial injury with secondary physiologic vascular effects could be acting together to affect the downstream capillary bed. Sympathetic activation in these patients could operate together with local vasoconstrictor responsiveness (41). Because not all patients have elevated blood pressure at the time of toxic reaction, however, hypertension is unlikely to be the sole mechanism for the toxic reaction process.
CT and MR images show transient brain abnormalities after seizure activity (58–61). Seizure activity usually is a symptom of an underlying brain disorder and is considered a consequence of brain irritation. During and immediately after seizure activity, certain regions of the brain typically are hyperperfused, as noted on single-photon emission CT (SPECT) perfusion studies (62). In the interictal period, local cerebral blood flow typically is diminished (62). Seizure activity, therefore, is associated with variations and alterations in blood flow in the brain. Whether such blood-flow changes lead to brain ischemia or infarction is not established. In addition, positron emission tomography studies have shown hypometabolism in the epileptogenic cortex with diminished fluorine 18-fluorodeoxyglucose activity in the interictal period (62). The regional blood flow variations could simply reflect changes in blood supply responding to metabolic needs during or after seizure activity (58, 62).
In the literature, many patients with CsA and FK-506 neurotoxic reactions have presented with seizures, but altered mentation or visual-field abnormality (without seizure activity) also is common (5, 7, 11, 17, 21). Six (27%) of 22 patients in our series had imaging changes that were characteristic of neurotoxic reaction, in the absence of seizure activity. Although the role of seizure activity is unclear, we suspect that neurologic problems, including seizures, were caused by the brain abnormalities present.
The time course of lesion development is an important and interesting feature in these patients. Lesions usually were noted on the initial image, but in some patients, lesions developed as a subacute process. Lesions typical of CsA neurotoxicity were seen in 10 (59%) of 17 patients who underwent CT first and in four (80%) of five patients who underwent MR imaging first. The initial CT scan was negative in four patients, but changes typical of CsA neurotoxicity then developed over an 8-d period; this development suggested a subacute process. In one patient, the CT scan obtained shortly after generalized seizures was normal but became floridly abnormal at 24-h follow-up (Fig 1). In seven (32%) of 22 patients, imaging was either normal (four patients) or nonspecific (three patients) at initial examination. The results included nonspecific findings on MR images obtained at initial presentation in one patient.
Nonspecific lesions may be noteworthy. Many patients receiving cyclosporin have tremulousness or nervousness, which may suggest CNS changes (25). Three patients in our study had nonspecific findings on initial images, but their 2-wk follow-up studies were positive for CsA toxicity. Nonspecific findings may be the initial brain changes that then develop into the lesions of CsA and FK-506 toxicity. Repeat or follow-up scanning in patients with CsA and FK-506 toxic reactions and normal or nonspecific brain imaging findings may be important in further characterizing the brain lesions and confirming the toxic process.
In two patients, CT scans showed lesion hemorrhage. In one additional patient, MR images depicted older hemorrhage along the inferior cerebellar surface. Two of these three BMT patients were conditioned with only cyclophosphamide and TBI. Acute hemorrhage appears to be an uncommon but important finding in CsA toxicity. Most patients undergoing allogeneic BMT are thrombocytopenic. Identification of acute bleeding with CT could affect the use of platelet transfusions.
The imaging features of eclampsia and chemotherapy toxicity have been noted to resemble the lesions in CsA toxicity (28, 51). We have seen several cases of chemotherapy toxicity that have a pattern that resembles CsA toxicity. Once again, the lesions encountered were within watershed zones; primarily, subcortical white matter abnormality exists, with sparing of the cortical gray matter.
Brain pathologic findings in two of our patients were reported as normal. Autopsy was performed 2 and 6 mo after toxic reaction in these two patients, and samples may have been obtained in only minimally involved regions. Several reports have described lesion resolution or improvement, even with extensive brain involvement (17, 18, 21, 23, 26, 28). Lesions in CsA neurotoxicity may not cause permanent parenchymal damage but may represent a transient alteration. Despite the absence of gross changes on external brain inspection, pathologists should be encouraged to obtain microscopic samples in the regions typically involved in CsA neurotoxicity and include histologic inspection of medium-sized blood vessels.
Conclusion
The lesion morphology of CsA and FK-506 neurotoxicity appears to depend on the conditioning regimen used before BMT. BMT patients conditioned with cyclophosphamide and chemotherapy (busulfan or thiotepa) have cortical and white matter involvement, with occasional cortical enhancement. Nontransplant patients and BMT patients conditioned with cyclophosphamide and TBI have only white matter involvement with no lesion enhancement. All lesions were located in a watershed distribution or in the anastomotic border zones. BMT-related TM was present in all patients examined, and vasculopathy may be the primary cause of CsA and FK-506 neurotoxicity. Edema with local mass effect is common, and acute hemorrhage is occasionally noted. Initial images in these patients may be normal or nonspecific at presentation (32% of our patients), but characteristic changes typically develop within 1–8 d.
Footnotes
1 Presented at the Annual Meeting of the European Neurological Society, Nice, France, June 1998.
2 Address reprint requests to Walter S. Bartynski, MD, Department of Radiology, The Western Pennsylvania Hospital, 4800 Friendship Avenue, Pittsburgh, PA 15224.
References
- Received June 18, 1999.
- Accepted after revision June 14, 2001.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Posterior reversible encephalopathy syndrome resulting from repeat bortezomib usage
- Neurotoxicology: Five new things
- Reversible Encephalopathy after Cardiac Transplantation: Histologic Evidence of Endothelial Activation, T-Cell Specific Trafficking, and Vascular Endothelial Growth Factor Expression
- Non-infectious manifestations of stem cell transplantation
- Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer
- Reversible diffusion MRI abnormalities and transient mutism after liver transplantation