
Abstract
BACKGROUND AND PURPOSE: Modifications of aneursym occlusion devices and other biologically active molecules may reduce the risk of recanalization by promoting vascular cell migration, adhesion, and proliferation. Our purpose was to apply in vitro methods in the qualitative and quantitative analysis of vascular smooth muscle cell (VSMC) interactions with collagen-impregnated microcoils.
METHODS: The adhesion of rat aortic VSMCs to collagen fiber bundles (CFBs), nitinol coils, and collagen-impregnated nitinol coils (CINCs) was examined by using an assay consisting of monopulse exposure to increasing concentrations of rat aortic VSMCs. Exposed devices were washed and examined by using confocal fluorescence microscopy. Adhesion coefficients, which quantitatively express the cell-binding quality of a surface, were determined by using a mathematical model for cell-device interactions.
RESULTS: VSMCs, attached to devices, spread out and extended cytoplasmic projections over the contact surface. Cell distribution was random on CFBs and within interloop troughs on nitinol coils. On collagen-impregnated coils, VSMCs were selectively concentrated on the collagen between coil loops. The average adhesion coefficient was 25.0 for CFBs, 8.5 for CINCs (250-μm pitch), and 6.5 for nitinol coils. Adhesion coefficient differences for the three devices were significant (P = .044).
CONCLUSION: The monopulse exposure assay is a simple and reproducible in vitro test that provides qualitative information about the morphology and topography of cell-device contacts and permits quantitative measurement of the intrinsic cell-binding quality of the test device. VSMCs exposed to collagen-impregnated microcoils selectively attach to collagen. Collagen enhances the rate of VSMC adhesion to embolic devices, and the degree of enhancement correlates with the surface area constituted by collagen.
Clinical studies (1, 2) have revealed that the rate of cerebral aneurysm recurrence after coil embolization therapy may be as high as 14%. Endovascular fibrosis may prevent aneurysm recurrence by mechanically stabilizing the endoluminal coil mass and by permanently obliterating the lumen with a stable connective tissue matrix that is resistant to recanalization. Embolization coils modified with collagen have been devised to augment vascular cell adhesion and fibrosis in embolized aneurysms (3–10). Some (3–7) have postulated that collagen is an effective substrate for vascular cell adhesion and that collagen effectively supports the migration and adhesion of vascular cells. Initial success with collagen coils has led to efforts to design a more effective bioactive embolic device by using novel biomaterials (5–7, 9, 11–14). The number of biomaterial preparations with theoretical potential is staggering, and new analytic tools that can provide rapid and meaningful results are required to narrow the scope of investigative efforts in this area.
Although long-term animal studies are necessary to completely elucidate the biological effects and practical limitations of any biomedical device, efforts to further advance the development of bioactive embolics would be facilitated by in vitro tests that rapidly provide information about cell-device interactions and that can be used to simultaneously screen large numbers of potentially useful biomaterials. Adequate in vitro characterization of cell-device interactions requires qualitative information about cell-device contacts and quantitative information about cell adhesion rates. In vitro screening would allow investigators to select a few of the most promising prototypes for further evaluation in animal models. This approach reduces the number of animals that would be sacrificed for in vivo testing, as well as the total cost of the required investigation, because animal studies are typically expensive. The following study was based on a model of in vitro analysis that we developed for this purpose.
Methods
Collagen Fibers and Collagen Fiber Bundles
Non–cross-linked type I collagen microfibers (40–50-μm diameter) were prepared from adult rat tail tendons. Collagen was dissolved by stirring freshly excised rat tail tendons in a solution of HCl (pH 2) for 10 hours. After centrifugation, the supernatant was filtered twice through Millipore membranes (0.65- and 0.45-μm pores), and NaCl was added to a concentration of 0.7 M. Collagen was precipitated at 4°C for 8 hours, pelleted by means of centrifugation, dissolved in HCl (pH 2), dialyzed, and pelleted again. The cycle of dissolution, dialysis, and centrifugation was repeated. The resulting collagen pellet was redissolved in HCl (pH 2) and sterile filtered. Crude microfibers were formed, as Kato et al described (15). The composition of each completed microfiber was more than 90% type I collagen, and the remaining mass was a mixture of type II and type III collagen (16).
Collagen fiber bundles (CFBs) were prepared by using a surgical microscope and sterile technique. In each case, six collagen microfibers were braided into a tightly packed bundle with locking microsurgical forceps and fastened at both ends with 10–0 Prolene sutures. CFBs were stored in sterile vials for later use in monopulse exposure assays.
Nitinol Coils and Collagen-Impregnated Nitinol Coils
Nitinol embolization coils were fabricated from a wire of nickel-titanium alloy (56% Ni, 44% Ti) with circular cross-section measuring 50 μm in diameter (Shape Memory Applications Inc, Santa Clara, CA). Straight microcoils with a 0.4-mm outer diameter and a helical pitch of 50 and 250 μm were created by winding nitinol wire on a mandrel and by means of heat setting at 550°C. In coils with a helical pitch of 50 μm, the loop of one coil turn was in contact with the subsequent coil loop, without an intervening space. In coils with a helical pitch of 250 μm, the loop of one coil turn was separated from the next loop by a distance of 200 μm.
All coils were gas sterilized with ethylene oxide and stored in sterile vials. Sterile coils were used for monopulse exposure assays, or they were impregnated with collagen for subsequent use in monopulse exposure assays.
Collagen-impregnated nitinol coils (CINCs) with a helical pitch of 250 μm were prepared by using a surgical microscope and sterile technique. In each case, six collagen microfibers were braided into a tightly packed bundle and pulled through the central cavity of a coil with a 10–0 Prolene suture on a straight needle.
Cell Cultures
Aortic smooth muscle cells from adult rats were isolated and cultured according to the method of Oakes et al (17). These cells were cultured in Dulbecco modified Eagle medium supplemented with l-glutamine, penicillin-streptomycin, and 10% fetal bovine serum. Established cultures were frozen and stored in liquid nitrogen for as long as 2 years. Subsequently, these cells were thawed and harvested for use in monopulse exposure assays after the fifth to eighth passages.
Monopulse Exposure Assays
Specimens of CFBs, nitinol coils, and CINCs with a helical pitch of 250μm were prepared and cut to a total length of 1.5 mm. Prior to cutting, devices containing collagen fibers were wet with serum supplemented media (Dulbecco modified Eagle medium with 10% fetal bovine serum) to prevent unraveling of the fibers. Three specimens of each device were individually placed into well inserts with a semipermeable polycarbonate membrane floor (Corning Costar Corp, Cambridge, MA). Each insert was 5 mm in diameter, with a pore size of 0.4 μm and a total capacity of 3 mL. The nine well inserts containing the specimens (three types of devices × three specimens of each device) were placed into wells on a single multiwell plate. Each specimen was immobilized in the center of its insert by applying 100 μL of serum-supplemented medium to its surface with a micropipette. A second multiwell plate was subsequently prepared in duplicate fashion (for trial 2).
Cultures of rat aortic smooth muscle cells were treated with trypsin, pelleted by means of centrifugation, and washed twice in serum-supplemented medium. Cell pellets were combined and suspended in serum-supplemented medium. Serial dilution was performed with the same medium to yield a series of three suspensions containing 2.6 × 106, 2.6 × 105, and 2.6 × 104 cells per milliliter.
Three-milliliter aliquots of each suspension were pipetted into the individual well inserts that contained each type of specimen. The wells surrounding the inserts were filled with serum-supplemented medium. This compartmental arrangement of the cells and media was used to concentrate the cells in a small volume surrounding the device while maximizing the gas exchange and total volume of medium available to the cells. The multiwell plates were placed on a rotoshaker (Barnstead/Thermolyne Corp, Dubuque, IA) and vigorously agitated for 30 minutes at 37°C. Incubation was continued for another 3.5 hours, without shaking, to extend the total cell-device interaction time to 4 hours, the approximate length of time required for cells to attach and spread on the surface of static tissue culture plates in our laboratory. The material in each well insert was then aspirated, and each of the specimens was washed three times with 3-mL volumes of serum-supplemented medium.
The washed specimens were subsequently transferred to fresh well inserts by using microsurgical forceps. The well inserts and surrounding wells were filled with serum-supplemented medium, and the plates were gently agitated on a rotoshaker at 37°C for 24 hours.
After 24 hours of incubation, all medium was aspirated from the well inserts and surrounding wells. Two mL of a live/dead cell fluorescent stain consisting of calcein AM and ethidium homodimer-1 (Molecular Probes Inc, Eugene, OR) was added to each well insert. After the multiwell plates were incubated at 37° C for 45 minutes, specimens were removed from the staining reagent, washed in phosphate-buffered saline, and transferred to glass coverslips for further analysis at confocal fluorescence microscopy with an argon laser (Carl Zeiss Inc, Thornwood, NY). Specimens were scanned to generate a series of four 6.3-μm-thick image sections. Each image section was a two-dimensional tangential cross-section of one of the four surfaces of the specimen: superior, inferior, and two lateral. The lateral surfaces of each specimen were examined last, after the specimen was rotated 90°.
We defined the fractional saturation (FS) as the percentage of the specimen’s surface area that was covered with viable cells. For each specimen, the FS was individually determined for each of its four surfaces by selecting a representative oval region of interest (ROI) on the corresponding image section. The ROI of each sample had a 400-μm-long axis, which was parallel to the long axis of the device, and a 300-μm short axis, which spanned the device transversely.
The surface area within the selected ROI was determined, and, by applying a series of masks to the image, the individual surface areas constituted by live cells and dead cells within the ROI were determined (Fig 1). Quantitative image analyses and surface-area determinations were performed with the LSM 510 Image Browser Program (Carl Zeiss Inc). Cells fluorescing red were counted as being dead, because red-fluorescent ethidium can penetrate and intercalate into only the nucleic acids of cells with damaged cytoplasmic membranes (18). Cells fluorescing green were counted as being viable, because only viable cells can enzymatically convert the non-fluorescent membrane permeant calcein AM to non-permeant green fluorescent calcein (18). The final FS (FSaverage) of each specimen, used to determine the adhesion coefficient, was calculated by averaging the individual FS values determined for each of its four surfaces.

Determination of the FS. Images are argon-laser fluorescence confocal micrographs of a CINC with a helical pitch of 250 μm obtained after monopulse exposure assay with an initial vascular smooth muscle cell (VSMC) concentration of 2.6 × 105 cells per milliliter (original magnification ×40).
A, An oval ROI, represented by the white line, is manually drawn on the device surface to begin the analysis. Wide solid straight arrows indicate loops of the nitinol coil; curved arrows, collagen fibers; thin solid straight arrow, live cells; open arrow, dead cells.
B, A mask is applied to the image matrix outside the ROI, and the surface area within the ROI is calculated.
C, The live cell surface area within the ROI is calculated by subtracting red fluorescence and masking photon-deficient pixels within the ROI.
Mathematic Model for Cell-Device Interactions
In the experimental system for the monopulse exposure assays, the adhesion of the cells to the device surface is a rate process that depends on the collision frequency between the cells and the device and on the intrinsic properties of the device surface that influence cell adhesion. The latter is expressed as the adhesion coefficient. In such experimental systems, only a fraction of collisions between the cell and the device surface result in successful cell adhesion. The adhesion coefficient is a derived parameter; it is a quantitative measure of the cell-binding quality of the surface of the test device. This measure is based on the assumption that all potential cell-binding sites on the surface of a device are available when cell-device interactions are initiated and that the entire surface of the device would be covered with cells if the interaction time was extended to infinity. The adhesion coefficient is proportional to the fraction of cell-device collisions that result in successful cell adhesion. The collision frequency between the cells and the device surface is proportional to the product of the concentration of available adhesion sites, or [AAS], on the device surface and the concentration of cells surrounding the device surface, or [VSMC].
According to our assumptions, cell adhesion to the device surface can be described by using the following rate expression: d[OAS]/dt = adhesion coefficient × [AAS] × [VSMC], where [OAS] is the concentration of occupied adhesion sites on the device surface, and d[OAS]/dt is its differential with respect to time, that is, the rate of cell adhesion to the device surface.
If we make the assumption that [VSMC] does not change substantially over the course of a given experiment, because [VSMC] is much greater than [AAS], a rearrangement of the original rate expression reveals that the adhesion coefficient expresses the rate of cell adhesion per binding site available on the device surface, as follows: (d[OAS]/dt ÷ [AAS])K = adhesion coefficient.
If [TAS] is the concentration of total adhesion sites on the device surface that are occupied and available, then [TAS] = [OAS] + [AAS]. If we substitute for [AAS] in the original rate expression, we find that d[OAS]/dt = adhesion coefficient ([TAS] − [OAS]) × [VSMC]0, where [VSMC]0 is the initial concentration of cells at time t = 0. By rearranging this equation to d[OAS]/([TAS] − [OAS]) = adhesion coefficient [VSMC]0dt and integrating from t = 0 to t = t and from [OAS] = 0 to [OAS] = [OAS], we have the expression ln {([TAS] − [OAS])/[TAS]} = −adhesion coefficient × [VSMC]0t. This expression can be rearranged to the following: [OAS] = [TAS] × (1 − e–adhesion coefficient[VSMC]0t). If we define FS as [OAS]/[TAS], rearrangement and substitution reveals that FS = 1 − e–adhesion coefficient[VSMC]0t. Further rearrangement demonstrates that ln (1 − FS) = −adhesion coefficient × [VSMC]0t. Consequently, a plot of ln (1 − FS) versus [VSMC]0t should yield a straight line with a slope of the negative of the adhesion coefficient.
The adhesion coefficients for each device were determined by plotting ln (1 − FS) against [VSMC]0t for each of two duplicate trials of the monopulse exposure assays. In a single monopulse exposure assay trial for a given device, three empirically determined FS values corresponded to three values of [VSMC]0: 2.6 × 106, 2.6 × 105, and 2.6 × 104 cells per milliliter. A fourth point (0,0) was added to each plot, because the FS and ln (1 − FS) must be zero when [VSMC]0 = 0. The raw adhesion coefficient were calculated as the absolute values of the slope of the best-fit line for each of these plots. Each final adhesion coefficient was calculated by multiplying the raw adhesion coefficients by 1010. This manipulation was performed to simplify the numeric results. The adhesion coefficients determined for each of two duplicate trials were averaged to obtain the final adhesion coefficient value reported for each type of device.
Statistical Methods
An analysis of variance (ANOVA) was used to determine the significance of differences in the adhesion coefficient among the three devices studied.
Results
Monopulse Exposure Assays
Examination of the CFBs with fluorescence confocal microscopy after monopulse exposure revealed a random surface distribution of the VSMCs, at all VSMC concentrations (Fig 2A). VSMCs that were attached to collagen fibers spread out over the fiber surface, with multiple cytoplasmic projections extending over the surface contours.

Distribution of VSMCs on the surface of embolic devices. Images are argon-laser fluorescence confocal micrographs of embolic devices obtained after monopulse exposure assays (original magnification ×40). Wide solid straight arrows indicate loops of the nitinol coil; curved arrows, collagen fibers; thin solid straight arrows, live cells; open arrows, dead cells.
A, CFBs exposed to an initial VSMC concentration of 2.6 × 104 cells per milliliter. VSMCs are spread over the convex surfaces of individual fibers and within the troughs between fibers.
B, Nitinol coils with a helical pitch of 50 μm exposed to an initial VSMC concentration of 2.6 × 105 cells per milliliter. VSMCs are preferentially distributed in the troughs between coil loops.
C, CINCs with a helical pitch of 250 μm exposed to an initial VSMC concentration of 2.6 × 105 cells per milliliter. VSMCs are focally concentrated on the surfaces of collagen fibers between consecutive coil loops.
In contrast, examination of nitinol coils revealed a preferential distribution of VSMCs in the troughs between consecutive coil loops, at all VSMC concentrations (Fig 2B). CINCs with a helical pitch of 250 μm were distinctly characterized by a marked concentration of VSMCs on the exposed collagen surfaces between coil loops (Fig 2C).
For each trial of monopulse exposure assay that we performed, the FSaverage at a specific VSMC concentration for each device is given in Table 1. The percentage standard deviation is reported as an estimate of the variability between the four sides of each device. For each VSMC concentration studied, the mean FS of each device is compared in Figure 3. Each mean FS value was calculated by averaging the eight empirically determined FS values from the two independent trials of a monopulse exposure assay.

Mean FS of the device surface with VSMCs. The mean FS for each of the three devices studied are compared at three initial VSMC concentrations. The calculated mean FS is presented in the upper left margin above its corresponding bar. Error bars indicate a single standard deviation above and below the mean (n = 8). CINC-250 indicates CINCs with a helical pitch of 250 μm; NiT-50, nitinol coils with a helical pitch of 50 μm.
A, Initial VSMC concentration of 2.6 × 106 cells per milliliter.
B, Initial VSMC concentration of 2.6 × 105 cells per milliliter.
C, Initial VSMC concentration of 2.6 × 104 cells per milliliter.
FSaverage values at specific VSMC concentrations for each device in each trial of a monopulse exposure assay
Adhesion Coefficient Determinations
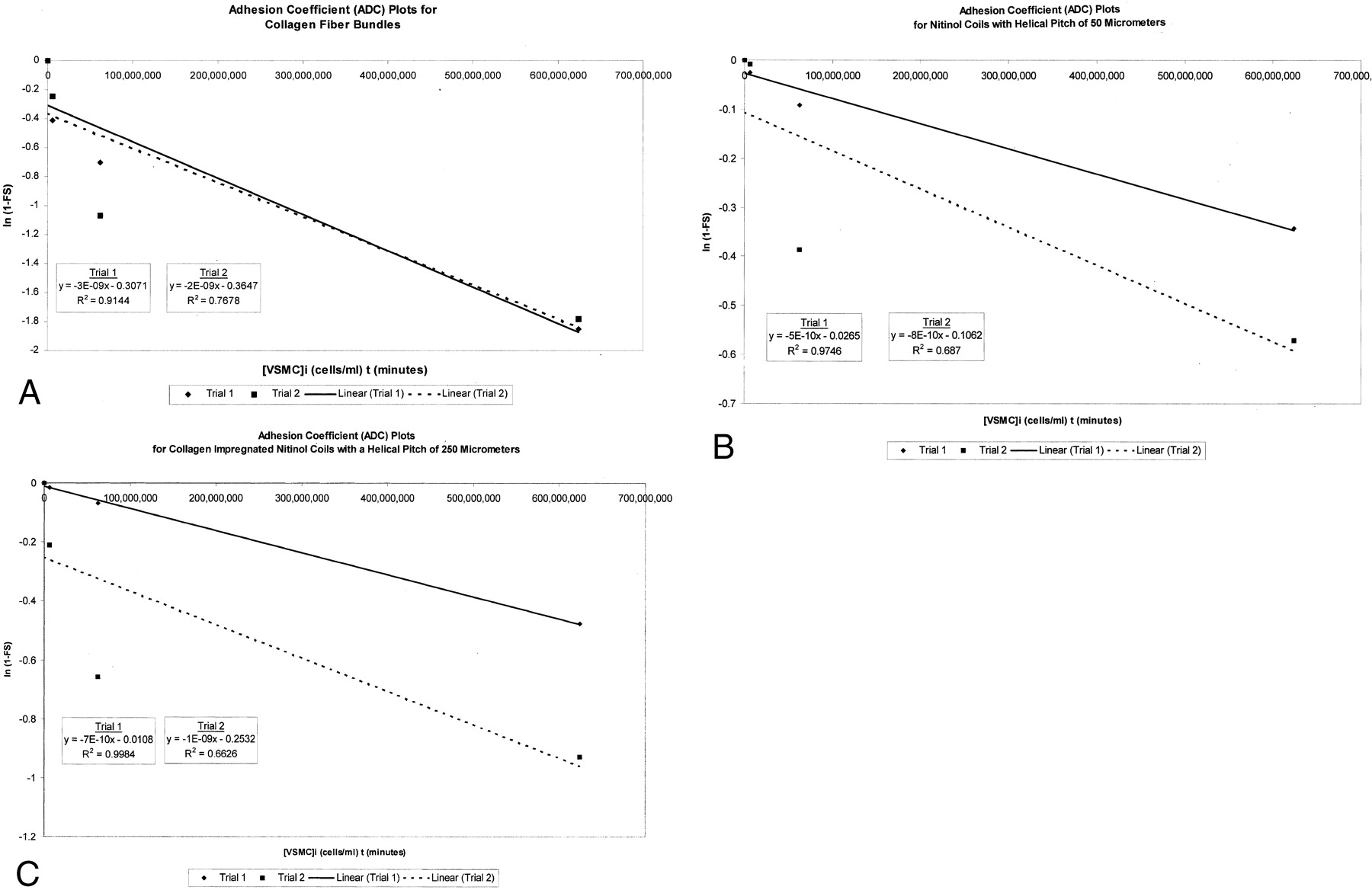
For each device, the adhesions coefficient plots for the two trials of the monopulse exposure assay are shown in Figure 4. The final adhesion coefficient values determined from these trials for each device are reported in Table 2.

Adhesion coefficient plots for two trials of the monopulse exposure assays are presented for each device. The plot for trial 1 is a cluster of four diamonds, with a solid best-fit line. Each plot for trial 2 is a cluster of four squares, with a dashed best-fit line. The equation that represents the best-fit line and the correlation coefficient for the scatterplot are shown. On each graph, ln (1 − FSaverage) is plotted against the product of 1) the initial concentration of VSMCs, or [VSMC]i, that was used to conduct the monopulse exposure assay and 2) the interaction time t of 240 minutes (4 hours).
A, Plots for CFBs.
B, Plots for nitinol coils with a helical pitch of 50 μm.
C, Plots for CINCs with a helical pitch of 250 μm.
Final adhesion coefficient for each trial of a monopulse exposure assay
Statistics
ANOVA revealed differences in the calculated adhesions coefficients for the three devices studied were statistically significant (P = .044).
Discussion
Monopulse Exposure Assays Provide Qualitative and Quantitative Data
In this study, monopulse exposure assays demonstrated the topographic distribution and morphology of VSMCs on the surface of collagen-impregnated microcoils at confocal fluorescence microscopy. On the basis of a first-order kinetics model, the adhesion coefficient, which is a quantitative measure of the cell-binding quality of a surface, was calculated for each device. The differences in the adhesion coefficients for a series of three devices, which were characterized by increasing levels of surface modification with collagen, were statistically significant (P = .044, ANOVA). This finding indicated that the adhesion coefficient can be used to distinguish embolic devices on the basis of their ability to support VSMC attachment.
Quantitative Analysis of Cell Adhesion Rate versus Number of Cells Attached
In contrast to other quantitative in vitro assays that are based on a determination of the number of cells, the monopulse exposure assay described in this report provides quantitative information about the cell adhesion rate per cell-binding site, with an assumption that all binding sites on the device surface are potentially available when cell-device interactions are initiated. Because the adhesion coefficient is normalized to the number of available binding sites, it allows comparison of devices with different surfaces areas (ie, different number of total potential cell-binding sites). Colorimetric tests, such as the microculture tetrazolium assay (MTA), can be used to determine the number of cells in vitro (19–22). Such tests, which rely on the mitochondrial bioreduction of a yellow tetrazolium salt to a purple formazan, do not provide morphologic or topographic information about cell-device contacts, and they do not enable direct determination of cell adhesion rates that are normalized to the number of available cell-binding sites on the surface of the test device (19–22). To determine the cell adhesion rate per cell-binding site, the FS of the device surface must be know. Although MTAs can be used to determine the FS, they require an estimate of the available binding sites on the surface of the test device. Such estimates can be empirically determined only with extended interaction times that are complicated by substantial cell-mediated surface modifications. Such surface modifications would be expected to promote additional cell adhesion and proliferation and to alter the measured adhesion coefficient so that it no longer reflects the intrinsic properties of the test device.
Kinetic Model
Monopulse exposure assays performed on CINCs with a 250-μm pitch revealed that VSMCs were selectively concentrated on exposed collagen surfaces between coil loops. The findings of these experiments indicate a definite preference of the VSMCs for collagen fibers instead of nitinol. The results are the basis of our kinetic model and quantitative analysis.
Because type I collagen fibers are a superior substrate for VSMC adhesion, the rate of adhesion of VSMCs to a nitinol device is inferred to be positively correlated with the device surface area constituted by collagen. If the adhesion coefficient is a true measure of cell-binding quality, it should also be positively correlated with the device surface area constituted by collagen. Results of a comparison of the different devices supports this relationship: CFB values were higher than those of CINCs with a 250-μm pitch, which were higher than those of nitinol coils.
The adhesion of VSMCs to the surface of extracellular matrix proteins, such as collagen, is a highly specific receptor-ligand interaction mediated by membrane-associated β1 integrins (23). The initial cell adhesion to metal-alloy surfaces, such as nitinol, probably involves nonspecific electrostatic interactions; however, specific integrin-mediated interactions are likely to play an important role as cells begin to deposit matrix on the metal surface (24). Although cell adhesion to bioartificial devices, such as collagen-impregnated coils, may involve a great deal of biochemical complexity, our experimental data conform well to simplified first-order kinetics. Semilog ADC plots for the three devices studied provided average correlation coefficients, in trials 1 and 2, of 0.830 for CINCs with a helical pitch of 250 μm pitch, 0.832 for nitinol coils, and 0.841 for CFBs. These results support the use of first-order kinetics to model cell-device interactions in monopulse exposure systems (Fig 4).
Experimental Design
Previous investigators (9) have reported on the interaction of endothelial cells with embolic devices in vitro. Although endothelial cells play an important role in the arterial response to injury and device implantation, the VSMC is the central mediator of tissue remodeling and repair in healthy and diseased arteries, and it is thought to be the principal cell type that mediates the vascular responses to device implantation (25–31). We used VSMCs to perform our in vitro studies because the response of VSMCs in vitro should most closely mimic the arterial response to device implantation in vivo.
Although platinum microcoils are used almost exclusively in clinical neuroendovascular therapy, we performed our studies by using nitinol coils, because the precise control of the helical pitch was an essential requirement in our experimental design. Unlike nitinol, platinum does not retain its original shape and pitch when it is deformed during the relatively traumatic process of collagen fiber insertion. Nitinol is a biocompatible alloy with a superelastic shape memory that has been widely used in the fabrication of implantable endovascular devices; it enabled us to easily fashion microhelices with a precisely defined pitch that remained relatively stable after collagen fibers were inserted (32–40).
We used type I collagen as the biomaterial modifier in our study because an abundance of data indicates that it can be used as a highly effective bioactive embolic agent (3–10, 41, 42). Compared with other naturally occurring extracellular matrix proteins, such as fibronectin and laminin, type I collagen most rapidly promotes the adhesion, migration, and growth of endothelial cells on coated embolic devices (9).
Effects of Device Modification with Collagen
Findings from animal studies (3–6, 8) with vein pouch aneurysm models suggest that embolic devices modified with collagen increase endoluminal fibrosis, and, therefore, they reduce the risk of aneurysm recanalization. The biologic phenomena underlying this effect are likely complex and multifactorial; they may involve the integrated activity of platelets, endothelial cells, adventitial fibroblasts, and VSMCs. Previous in vitro investigations (9) have shown that endothelial cells grow on stationary embolic devices more rapidly when the devices are coated with type I collagen. Our in vitro data show that the modification of devices with type I collagen enhances the rate of VSMC adhesion to the device and that cell adhesion preferentially occurs in areas of exposed collagen. These results are supported by those of other investigators (43, 44) who have found that soluble, monomeric type I collagen is a potent VSMC chemotaxin and that fibrillar type I collagen strongly promotes the adhesion of smooth muscle cells.
Conclusion
The results of this study indicate that the monopulse exposure assay is a valid and reproducible in vitro method that provides 1) qualitative information about the morphology and topography of cell-device contacts and 2) quantitative information about the rate of vascular cell adhesion per cell-binding site.
Monopulse exposure assays demonstrate that VSMCs exposed to collagen-impregnated microcoils selectively attach to collagen microfibers. Quantitative analysis reveals that modification of the microcoils with collagen enhances the VSMC adhesion rate and that the degree of enhancement is positively correlated with the device surface area constituted by collagen. These findings are consistent with previously published in vitro and in vivo data (3–7, 9, 10, 42).
Device modifications, which enhance the rate of VSMC adhesion, may increase fibrosis and luminal obliteration of aneurysms after endovascular therapy. Therefore, the selection of biomaterials for the modification of embolic devices can be guided with monopulse exposure assays to allow investigators to rapidly screen large numbers of biomaterial preparations in vitro, before they commit to expensive long-term animal studies.
Acknowledgments
We are grateful to Steven Woodard, MS, for his guidance and support in all aspects of our confocal microscopy work. We thank Linda Donoff for her assistance in preparation of the manuscript.
Footnotes
This study was funded in part by the 1999 Sterling Radiological Society of North America Scholar Award (H.J.C.) and a grant from the Emory University/Georgia Institute of Technology Biomedical Research Center.
References
- Received March 8, 2001.
- Accepted after revision October 8, 2001.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}