
Lysosomal free sialic acid storage diseases are rare inborn errors of metabolism with autosomal recessive inheritance that are caused by a defect in the lysosomal membrane-specific carrier for sialic acid and uronic acids (1). The gene, SLC17A5 (MIM 604322), localized on chromosome 6q14-q15 (2), has recently been identified and sequenced (3). The defective egress of free sialic acid from the lysosome leads to both an excessive store of sialic acid in many tissues and considerable excretion in the urine (4). Cases are classified into two different phenotypes that share the same biochemical abnormality, the most severe being the infantile sialic acid storage disease (MIM 269920) and the mild one being Salla disease (MIM 604322). We herein report a case of infantile sialic acid storage disease studied with serial cranial sonography and MR imaging during the first months of life.
Case Report
The female patient was born at 36 weeks gestational age to healthy, young, unrelated parents. Cesarean section was performed because of fetal distress, microcephaly, and anhydramnios. During pregnancy, sonography performed at 30 weeks of gestational age showed subcutaneous edema, hydrops, and lateral ventricle enlargement. The results of karyotype analysis of amniotic cells were normal. At birth, the Apgar score was 7/9 and weight was 2200 g. Ascites, petechiae, hepatosplenomegaly, and dysmorphic features (coarse facies with hyperthelorism, depressed nasal bridge, periorbital edema, and full cheeks) were present. Hypertrophic cardiomyopathy and nephrotic syndrome were evident immediately after birth. Subsequently, the patient did not acquire any psychomotor skill and died at 6 months of age for cardiac failure in a context of refractory to treatment nephrotic syndrome. Diagnosis of infantile sialic acid storage disease was made based on the finding of increased free sialic acid in the urine (sialic acid, 4068 μg/mg creatinine; normal value is 150–300 mg/dL; free sialic acid, 83%). Molecular analysis of the patient DNA showed a homozygous GT deletion (position 1138-1139) in exon nine of SLC17A5 gene. This mutation causes a frame shift that probably leads to the synthesis of a truncated protein. The patient underwent serial transfontanellar sonographic studies performed at the ages of 2, 21, and 132 days of life. The sonograms were obtained by 7.5-MHz transducers. MR imaging was performed at 50 days of life with a 1.5-T system obtaining fast spin-echo T2-weighted and spin-echo T1-weighted sequences. The sonography performed on the 2nd day of life showed mild hyperechogenicity of the periventricular white matter and thalami (Fig 1A) and corpus callosum hypoplasia. The follow-up study performed at 21 and 132 days of life showed progressively increased evidence and wideness of parenchymal echogenicity (Fig 1B). No ventricular dilation was detected by any of the examinations, except for a very slight enlargement of the left lateral ventricle. On MR images, normal white matter myelination was absent. The posterior limb of internal capsule, almost partly myelinated at birth, showed abnormal signal intensity on both T2- and T1-weighted images. On the contrary, the periventricular white matter showed symmetric slight hypointensity on the T2-weighted sequence (Fig 2A) and hyperintensity on the T1-weighted sequence (Fig 2B). This aspect was more evident in the frontal region. The MR examination confirmed the extreme hypoplasia of the corpus callosum; marked thinning of chiasm and optic tracts was also evident (Fig 2C).

Sonographic evaluation.
A, Sagittal view sonogram obtained at 2 days of life.
B, Sagittal view sonogram obtained at 132 days of life. A progressive increase of parenchymal echogenicity is shown.
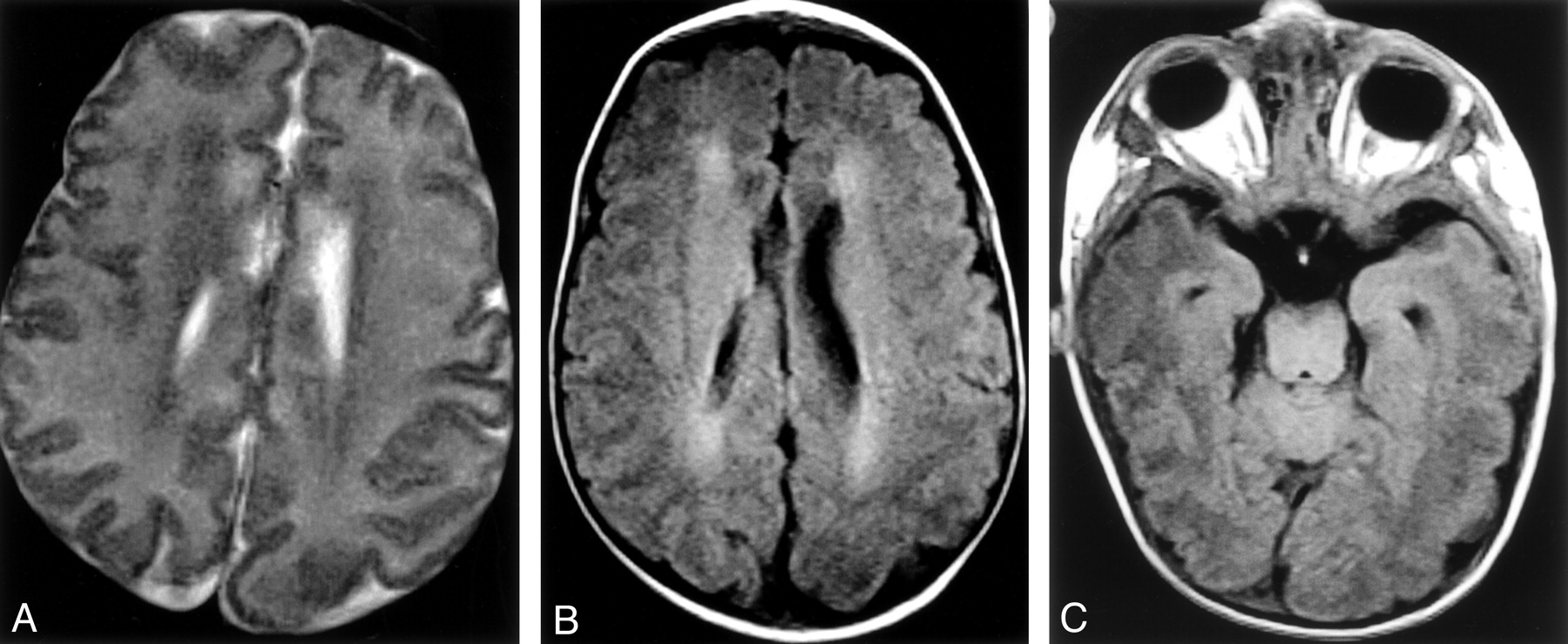
MR imaging study.
A, Axial view T2-weighted MR image. Periventricular white matter shows symmetric hypointensity on the T2-weighted sequence.
B, Axial view T1-weighted MR image. Periventricular white matter shows hyperintensity on the T1-weighted sequence.
C, T1-weighted MR image shows thinning of the optic chiasm.
Discussion
Infantile sialic acid storage disease is a severe rare inborn lysosomal storage disorder caused by defective transport of free sialic acid through the lysosomal membrane (4). The storage of free sialic acid is found in many tissues, including skin, kidney, liver, cardiac muscle, and brain (5, 6). Patients present with hydrops fetalis and dysmorphic features; hepatosplenomegaly is evident at birth. Hypertrophic cardiomyopathy and nephrotic syndrome are very often associated. Survival of these patients is <2 years (4). No treatment is presently available. Prenatal diagnosis is possible during the second trimester of pregnancy by biochemical analysis of free sialic acid in amniocytes and amniotic fluid or during the first trimester by molecular analysis, provided the mutations of the gene in the family are known. Until now, approximately 27 cases of infantile sialic acid storage disease have been described (4) but no information regarding the associated cerebral MR imaging is available. On the contrary, the neuropathologic findings are reported. Severe dysmyelination of the white matter, gliosis, and microcalcifications have been noted, and hypoplasia of the corpus callosum is evident. Microscopic examination of hematoxylin and eosin-stained brain tissue sections shows large numbers of vacuoles, consistent with lysosomes within neurons, and astrocytes give their cytoplasm a foamy to clear appearance (4, 6).
In our case, both cranial sonography and MR imaging showed an abnormal appearance of the periventricular white matter, which was characterized, respectively, by increased echogenicity and signal intensity alteration on T1- and T2-weighted sequences. The neuropathologic changes involving the cytoplasm of brain cells might explain these findings. A marked thinning of the corpus callosum, in agreement with the neuropathologic data reported in the literature, was also evident based on both sonograms and MR images and probably reflects a depletion of myelinated fibers that cross the midline. In addition, the MR imaging examination was able to show the absence of the normal myelination of the white matter at birth, consistent with a prenatal arrest of myelination. Normal signal intensity of myelination was seen only in the dorsal part of brain stem; the cerebellar and cerebral white matter was unmyelinated. On MR images, we also noted a thinning of chiasm and optic tracts, indicating an involvement of the optic pathways in this disorder. The main differential diagnosis of these MR imaging findings included lysosomal storage disorders and Pelizaeus-Merzbacher disease (mainly because of the failure of myelination), but the clinical findings and the sex of the patient did not support this diagnosis. Sonograms of our patient showed progressive increase of echogenicity of periventricular and subcortical white matter involving thalami and basal ganglia. The differential diagnosis of this finding at the beginning included perinatal asphyxia. This hypothesis could be ruled out on the basis of the increased diffusion of echogenicity with time and the absence of necrotic signs (periventricular leukomalacia). In addition, the clinical history of the patient indicated considering the possibility of a storage disease. In our case, cerebral sonography detected abnormal imaging in infantile sialic acid storage disease as early as 2 days of life, showing the progression of the disease over a period of a few days. We think that echography is a useful, safe, and manageable way of monitoring progression of disease in this and probably other severe lysosomal storage disorders during the first period of life when the anterior fontanel is still patent. MR imaging, because of its high anatomic detail and contrast resolution, may provide more information, mainly regarding myelination and white matter alterations. Although not specific, both sonography and MR imaging can support the diagnosis of infantile sialic acid storage disease made based on clinical and biochemical findings.
- Received April 10, 2002.
- Accepted after revision August 12, 2002.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}