
Abstract
Summary: We present brain MR images in three patients with neonatal-onset hyperammonemic encephalopathy resulting from urea-cycle disorders (two sisters with deficiency of the carbamyl phosphate synthetase I reaction step and one boy with an ornithine transcarbamylase deficiency). MR imaging revealed almost identical findings of injury to the bilateral lentiform nuclei and the deep sulci of the insular and perirolandic regions; to our knowledge, this pattern has not been previously reported. We hypothesize that these lesions presumably reflect the distribution of brain injury due to hypoperfusion secondary to hyperammonemia and hyperglutaminemia in the neonatal period.
The urea cycle incorporates excess nitrogen into urea, a water-soluble waste product, preventing the accumulation of toxic nitrogenous metabolites in the body. Five well-documented urea cycle disorders have been described, each representing a defect in the catalytic efficacy of one of the enzymes of the cycle (1). Although brain MR imaging findings in late-onset types have been described (2–9), few reports describe the neuroimaging results in neonatal-onset cases (10, 11). We herein describe the clinical presentation and brain MR imaging findings in three patients with neonatal-onset urea-cycle disorders. The patients were two sisters with deficiency of the carbamyl phosphate synthetase (CPS) I reaction step and one boy with an ornithine transcarbamylase deficiency (OTCD). The images revealed almost identical findings in the bilateral lentiform nuclei and in the perirolandic and insular cortices.
Case Reports
Case 1
This girl, aged 20 months at the time of this report, was the third child born to healthy young nonconsanguineous parents, and the older sister of the patient in case 2. The patient had no known history of in utero exposure to infections, drugs, or other teratogens. She was vaginally delivered at term without complications. She presented at the age of 2 days with lethargy, anorexia, and respiratory distress, progressing to coma, generalized tonic-clonic seizures, and the need for ventilatory support. Elevated serum ammonia levels as high as 1700 μmol/L (normal range, 64–107 μmol/L) were found. Electrolyte levels and liver function results were normal, with negative blood and CSF cultures. Quantitative serum amino acid analysis revealed the following values: glutamate, 362 μmol/L (normal range, 0–50 μmol/L); glutamine, 1268 μmol/L (normal range, 538–958 μmol/L); citrulline, 0 μmol/L (normal range, 8–29 μmol/L); arginine, 26 μmol/L (normal range, 22–88 μmol/L); and ornithine, 47 μmol/L (normal range, 49–151 μmol/L). Argininosuccinate was undetectable, and urinary orotate was 1–2 μmol/mol Cr (normal range, 0–5 μmol/mol Cr). Urine organic acid analysis revealed no specific abnormalities. DNA analysis showed no mutation in the OTC gene. A diagnosis of deficiency of CPS I reaction step was suspected. After 40 hours of hemodialysis and medical therapy (intravenous sodium benzoate, sodium phenylacetate, and arginine hydrochloride), the patient’s serum ammonia level normalized to 50 μmol/L. At the age of 2 weeks, an MR study was performed with a 1.5-T superconducting magnet (GE Medical Systems, Milwaukee, WI). Spin-echo T1-weighted images (TR/TE/NEX, 500/16/2) of her brain demonstrated T1 shortening in bilateral lentiform nuclei (globi pallidi more than putamina) and the deep sulci of insular and perirolandic regions (Fig 1A and C). Spin-echo T2-weighted images (3000/120/1) showed bilateral low signal intensity in the globi pallidi and high signal intensity in the corpora striata and white matter subjacent to the insular and perirolandic cortices (Fig 1B). Intraventricular hemorrhage and retrocerebellar subdural hematoma were also identified. At age 7 months, after two subsequent episodes of significant hyperammonemia, the patient’s brain MR images showed volume loss, most notably in the basal ganglia. In addition, the images depicted T1 shortening in the putamen and diffuse cortical atrophy (Fig 1D). T2-weighted images showed T2 prolongation in the lentiform nuclei. Myelination was slightly delayed for her age. The patient underwent cadaveric liver transplantation at the age of 8 months. Since the transplantation, she has been asymptomatic but has truncal hypotonia, appendicular hypertonia, choreoathetosis, and global neurodevelopmental delays. This aggregate clinical picture was consistent with the neuroimaging findings.
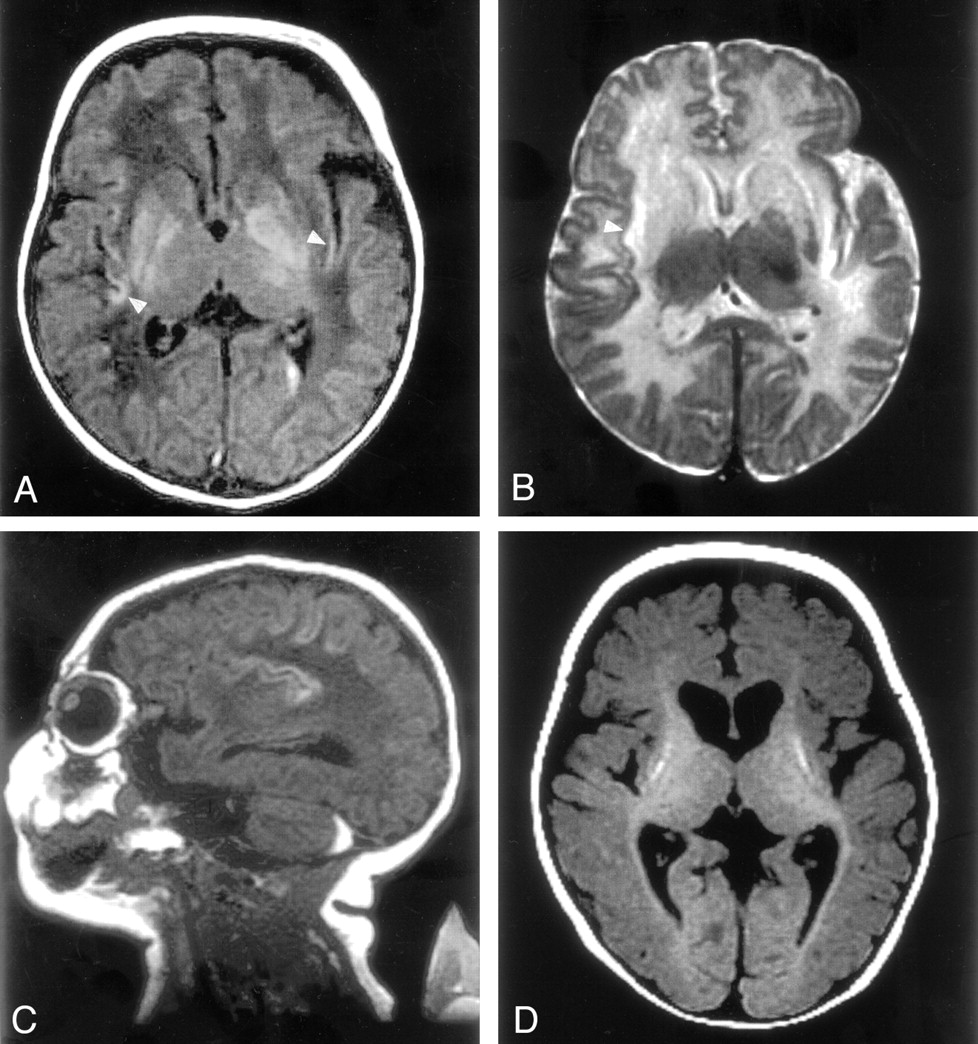
MR images in case 1.
A, T1-weighted image (500/16/2) of the brain demonstrates T1 shortening in the bilateral lentiform nuclei (globi pallidi more than the putamina) and insular cortex (arrowheads). Left intraventricular hemorrhage is recognized.
B, T2-weighted image (3000/120/1) shows low signal intensity in the left globus pallidus and high signal intensity in the corpora striata and white matter subjacent to the insular cortex (arrowhead).
C, Sagittal T1-weighted image shows T1 shortening in the deep sulci of the insular and perirolandic regions and a retrocerebellar subdural hematoma.
D, T1-weighted image obtained at the age of 7 months shows volume loss in the basal ganglia with T1 shortening in the putamina, reduced volume of cerebral white matter, and diffuse cortical atrophy. Myelination was recognized in the posterior limb of the internal capsule and in the optic radiation.
Case 2
The second girl, aged 3 months at this writing, was the younger sister of the patient in case 1. Her mother’s pregnancy was marked by late prenatal care and treatment for positive vaginal GBS culture. Like her older sister, this patient presented with lethargy, anorexia, and vomiting at the age of 2 days. She required intubation because of respiratory distress, and anticonvulsant therapy was started with the onset of clonic arm jerking. Her serum ammonia concentration increase to 978 μmol/L, with simultaneous urine orotate levels ranging from 1 to 2 μmol/mol Cr. Serum amino acid analysis revealed these levels: glutamine, 1710 μmol/L; citrulline, 0 μmol/L; arginine, 24 μmol/L; and ornithine, 48 μmol/L. Argininosuccinate was undetectable. Urine organic acid analysis revealed no specific abnormalities. The patient’s ammonia level normalized after 34 hours of hemodialysis and medical therapy. She showed gradual neurologic improvement. DNA analysis showed no mutation in the OTC gene. Hepatic CPS I enzyme activity was 20% the control value; this finding was consistent with the diagnosis of a deficiency of CPS I reaction step.
At the age of 2 weeks, T1-weighted images showed T1 shortening in the bilateral lentiform nuclei (the globi pallidi more than the putamina) and the deep sulci of insular and perirolandic regions. T2-weighted images demonstrated high signal intensity in the medial putamina, and white matter subjacent to insular and perirolandic cortices, as well as heterogeneous high and low signal intensity in the globi pallidi. These imaging characteristics were almost identical to those of case 1, as studied at age 2 weeks. At the age of 6 weeks, the patient in case 2 underwent cadaveric liver transplantation and, with the exception of non-oral food intake, she displayed age-appropriate development.
Case 3
This boy, 3 years old at this writing, was the first child born to healthy young nonconsanguineous parents. His mother’s pregnancy was uncomplicated, and delivery was by means cesarean section because of his breech presentation at term. The patient presented with lethargy, vomiting, and generalized tonic seizures at the age of 4 days. Elevated serum ammonia levels as high as 1420 μmol/L were found, with normal routine laboratory test results. The serum ammonia level normalized after 2 weeks of intermittent multiple rounds of hemodialysis plus medical therapy. DNA analysis revealed a missense mutation (Ser-90-Asn) in the OTC gene, confirming the diagnosis of OTCD.
At the age of 2 weeks, T1-weighted images showed T1 shortening in the bilateral lentiform nuclei (the globi pallidi more than the putamina) and the deep sulci of posterior insular and perirolandic regions. T2-weighted images demonstrated high signal intensity in the caudate head, medial putamina, and white matter subjacent to the insular and perirolandic cortices, as well as heterogeneous high and low signal intensity in the globi pallidi. These findings were almost identical to those of the previous two cases. This patient underwent cadaveric liver transplantation at the age of 7 months. After transplantation, he had been asymptomatic, but he had moderate neurodevelopmental delays.
Discussion
Urea-cycle disorders are important causes of neonatal metabolic encephalopathy. Affected patients are healthy at birth, but they later manifest signs and symptoms of encephalopathy related to the accumulation of urea-cycle metabolites, including ammonia and glutamine. Full-term neonates, with no obstetric risk factors, appear healthy for 24–48 hours and then exhibit progressive lethargy, hypothermia, and apnea, accompanied by high blood ammonium levels. Patients with milder cases can present with encephalopathy (eg, vomiting, abnormal mental status, ataxia, seizures, or developmental delay) at any age from infancy to adulthood.
The pathophysiologic mechanism of central nervous system injury in urea-cycle disorders is not completely understood. One theory invokes the intracerebral accumulation of glutamine as the major cause of the encephalopathy (1). The presence of high levels of ammonia results in the conversion of large amounts of glutamate to glutamine by glutamine synthetase; this occurs mainly in astrocytes. The accumulation of large quantities of glutamine is thought to cause changes in intracellular osmolality and result in subsequent astrocyte swelling, brain edema, intracranial hypertension, and cerebral hypoperfusion. In support of this theory, some have demonstrated that the cerebral edema associated with hyperammonemia can be prevented by impeding glutamine accumulation in the brain, suggesting that hyperammonemia is necessary but not sufficient to produce cerebral edema (1). This theory is consistent with the confirmation of high glutamine concentrations in patients with hyperammonemic encephalopathy with proton MR spectroscopy results (3, 12) and with the presence of extraordinarily high cerebrospinal concentrations of glutamine in OTCD patients with hyperammonemic encephalopathy.
The MR images in our three cases revealed almost identical findings in the bilateral lentiform nuclei and in the perirolandic and insular cortices; to our knowledge, these occurred in a pattern not previously reported. Previous reports in the literature (2–9) represent mostly late-onset urea-cycle disorders. Although the findings in these reports are varied, we were able to divide them into four common patterns: type 1, diffuse severe cerebral edema followed by diffuse atrophy (10, 11); type 2, extensive infarct-like abnormality, often presenting as acute hemiplegia (2–6, 11); type 3, presumably ischemic lesions in cerebral intervascular boundary zones (4, 5, 7); and type 4, reversible symmetric cortical involvement of the cingulate gyri, temporal lobes, and insular cortex with sparing of the perirolandic cortex (8, 9). The clinical severity (in decreasing order of severity) and the ages at onset are as follows: type 1, neonatal period or infancy; types 2 and 3, infancy to childhood; and type 4, adulthood. Symmetric cystic lesions at the gray matter-white matter junction, especially in the sulcal depth of the frontal, parietal, hippocampal, and insular regions, are known early pathologic lesions resulting from hyperammonemic encephalopathy (13, 14). Such perisulcal lesions are strongly suggestive of diminished cerebral perfusion in the setting of elevated intracranial pressure (15), which is typical in hyperammonemic encephalopathy. We propose that, on MR images, type 4 lesions might reflect such mild and partly reversible perisulcal ischemic insults. Therefore, we speculate that, in urea-cycle disorders, type 1, 3, or 4 lesions result from the same condition of hypoperfusion during hyperammonemia and that differences in residual enzyme activity underlie the variations in the age of onset and in the clinical severity. However, the pathophysiology of the type 2 lesion remains unclear because MR angiography and conventional angiograms fail to show any abnormality (3).
Despite the different causes of hyperammonemia, the patterns of subacute injury with primary involvement of the bilateral lentiform nuclei and the deep sulci of the perirolandic and insular regions was nearly identical. This observation suggests that the pattern detected with MR imaging reflects injury due to hyperammonemia and related toxicity, irrespective of the specific underlying proximal urea-cycle disorder. The pattern of primary cortical involvement resembled type 4 lesions; however, it obviously differed in the involvement of the perirolandic cortex and lentiform nuclei. Consistent with the MR findings in these patients, the histopathologic results in neonatal-onset urea-cycle disorders demonstrate cortical neuronal loss and spongiform changes at the gray matter-white matter junction far more frequently in the insula and also in the basal ganglia (1, 13, 14).
What is the mechanism of injury? In general, the most metabolically active and mature regions of the premature brain, those that have the most advanced myelination, perfusion and glucose uptake, are the regions that are damaged first in the setting of nearly complete cessation of brain perfusion (16). At 40 weeks after conception, the basal ganglia and perirolandic region have the highest metabolic activity; this is in addition to the brain stem and thalamus, which also have high activity beginning in the third trimester (16). Therefore, we might reasonably speculate that the MR images in our cases reflect injury to those regions most susceptible to hypoperfusion in the neonate under hyperammonemic conditions. We can also postulate that longer and more severe hypoperfusion due to hyperammonemia could lead to type 1 lesions (diffuse severe cerebral edema followed by diffuse atrophy), as previously reported (10, 11). In neonates, brain injury from a urea-cycle disorder can be differentiated from that due to profound hypotension by the absence of thalamic injury (characteristic in profound hypotension in neonates) and by normal neurologic status within the first few days of life. Asphyxiated neonates typically have abnormal findings from the time of delivery, with seizures starting in the first 24 hours after birth.
A prolonged hyperammonemic coma is associated with impairment of intellectual function and substantial parenchymal injury in patients who survive neonatal hyperammonemic encephalopathy (17). Thus, early diagnosis and treatment may prevent chronic impairment. Despite therapy with all treatment options (except for liver transplantation), twin patients with neonatal-onset deficiency of the CPS I reaction step had subsequent neurologic deterioration (associated with a type 1 lesion) after subsequent episodes of hyperammonemic encephalopathy (10). Accordingly, we would suggest that early liver transplantation should be considered in patients with neonatal-onset urea-cycle disorders. Knowledge of the MR findings may help to expedite the diagnosis and treatment.
Conclusion
We herein described the brain MR imaging findings in three patients with neonatal-onset urea cycle disorders: two sisters with a deficiency of the CPS I reaction step and one boy with OTCD. The MR images revealed almost identical findings in the bilateral lentiform nuclei and in the perirolandic and insular cortices, a pattern that has not been reported previously, at least to our knowledge. We speculate that the MR imaging results in our cases reflect injury to those regions most susceptible to hypoperfusion in the neonate under hyperammonemic conditions. Knowledge of the MR findings may help to expedite the diagnosis and treatment of neonatal-onset urea-cycle disorders. We also reviewed and discussed the previous neuroradiologic findings in urea-cycle deficits divided them into four common patterns.
Acknowledgments
The authors wish to thank the patients and their families for their contribution to this study.
Footnotes
This work was supported in part by funding from the Niemann-Pick Foundation, the California Genetic Disease Branch, and NIH grant M01RR01271 to the UCSF Pediatric Clinical Research Center.
References
- Received August 12, 2002.
- Accepted after revision October 20, 2002.
- Accepted after revision October 20, 2002.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
Jump to section
Related Articles
Cited By...
- The incidence of movement disorders increases with age and contrasts with subtle and limited neuroimaging abnormalities in argininosuccinic aciduria
- The Perirolandic Sign: A Unique Imaging Finding Observed in Association with Polymerase {gamma}-Related Disorders
- Teaching NeuroImages: MRI findings in carbamoyl phosphate synthetase 1 deficiency
- Acute Hyperammonemic Encephalopathy in Adults: Imaging Findings