
Abstract
Summary: Vascular and cortical anomalies have been found in a family with parietal foramina type 2 (PFM2), which is determined by the ALX4 gene. It is believed that ALX4 has a bone-restricted expression. We report a case of PFM with age-related size variation in a 4-year-old boy, as well as in his mother, aunt and grandfather. MR imaging of the child demonstrates prominent malformations of cortical (polymicrogyric cortex with an unusual infolding pattern) and vascular development (persistence median prosencephalic vein), associated with high tentorial incisure periatrial white matter changes.
Parietal foramina (PFM; Online Mendelian Inheritance in Man accession number 168500) is a skull ossification disorder with symmetrical oval defects in the parietal bones separated by a narrow bone bridge. The size of the parietal foramina decreases with age, and intrafamilial variability is observed (1, 2). Heterozygous mutations of MSX2 homeobox gene (5q34–35; 3) and ALX4 homeobox gene (11p11–12; 4, 5) determine classification as PFM type 1 (PFM1) or PFM type 2 (PFM2). These mutations result in functional haploinsufficiency critical for human skull ossification, and to date PFM is known as a normal skull variant, not related to other abnormalities.
We describe malformations of vascular and cortical development with distinct degrees of severity in one affected family of PFM2 with an identified ALX4 homeodomain transcriptor factor mutation (653G→A encoding Arg218Gln). Molecular studies of this family were previously reported by Mavrogiannis et al (6). Scant information is available on intracranial PFM-related anomalies (7, 8), and to the best of our knowledge, no information is available on a stated genetic mutation and familial study demonstrating severity variability.
Case Reports
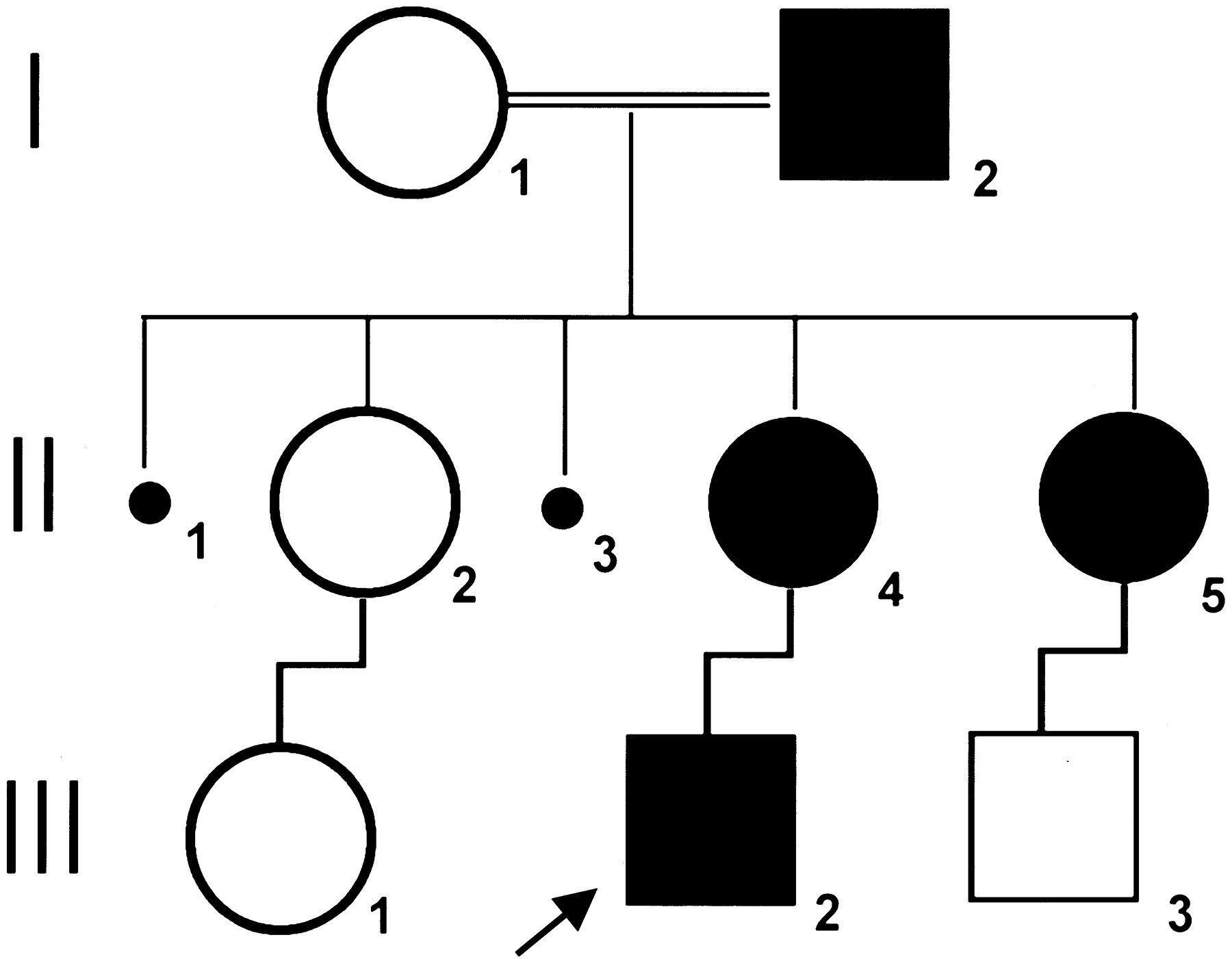
The patient (III-2, a 4-year-old boy) was born at term by caesarian section after an uneventful pregnancy. Physical examination, at the age of 3 months, showed a large bone defect (PFM). During follow-up, physical and neurologic examinations consistently revealed normal neurologic development and physical growth. His mother (II-4, 22 years old), aunt (II-5, 25 years old), and grandfather (I-2, 55 years old) were also found to have radiologically documented symmetrical PFM, although smaller than that of III-2. Figure 1 represents the pedigree of the family.

Pedigree of the affected family.
Clinical history, neurologic examination, and neuroimaging evaluation (skull radiographs and CT scan of the head with 3D cranial vault reformations) of this family were obtained, including an interval study of the child (at 13 months and 26 months). The unusual intracranial features observed by CT analysis led to prospective MR imaging, MR venography, and neurophysiological investigation in patients I-2, II-4, and III-2. Patient II-5 refused further analysis.
The neuroimaging protocol included helical CT of the head (with postprocessing 3D views of the cranial vault), structural MR imaging at 1.5T (three orthogonal planes with conventional spin-echo pre- and postintravenous paramagnetic contrast medium administration), and MR venography (time of flight).
Neuroimaging Studies
Conventional radiographs showed characteristic calvarial bone defects consistent with PFM. These findings were better assessed by cranial CT imaging in three of these patients, and an unusual tentorial and paramedian occipital region subjacent to the bony defects was demonstrated. The main MR imaging findings presented in all cases were as follows (Fig 2):
Polymicrogyric boundaries located mainly on the mesial portion of the occipital lobes;
Vascular embryonic pattern (9) characterized by persistent median prosencephalic vein with an underdeveloped straight sinus;
High insertion of the tentorium cerebri with an enlarged posterior fossa; and
Deep white matter signal intensity changes (T1 hypointensity but T2 and fluid-attenuated inversion recovery hyperintensity) of the posterior periatrial regions, bilaterally.

Neuroimaging studies of the affected family (III-2, II-4, and I-2). CT scan of the head with 3D cranial vault reformations (CT3D); axial plane MR imaging without contrast enhancement (T1-weighted MR imaging) and MR venography (MRv).
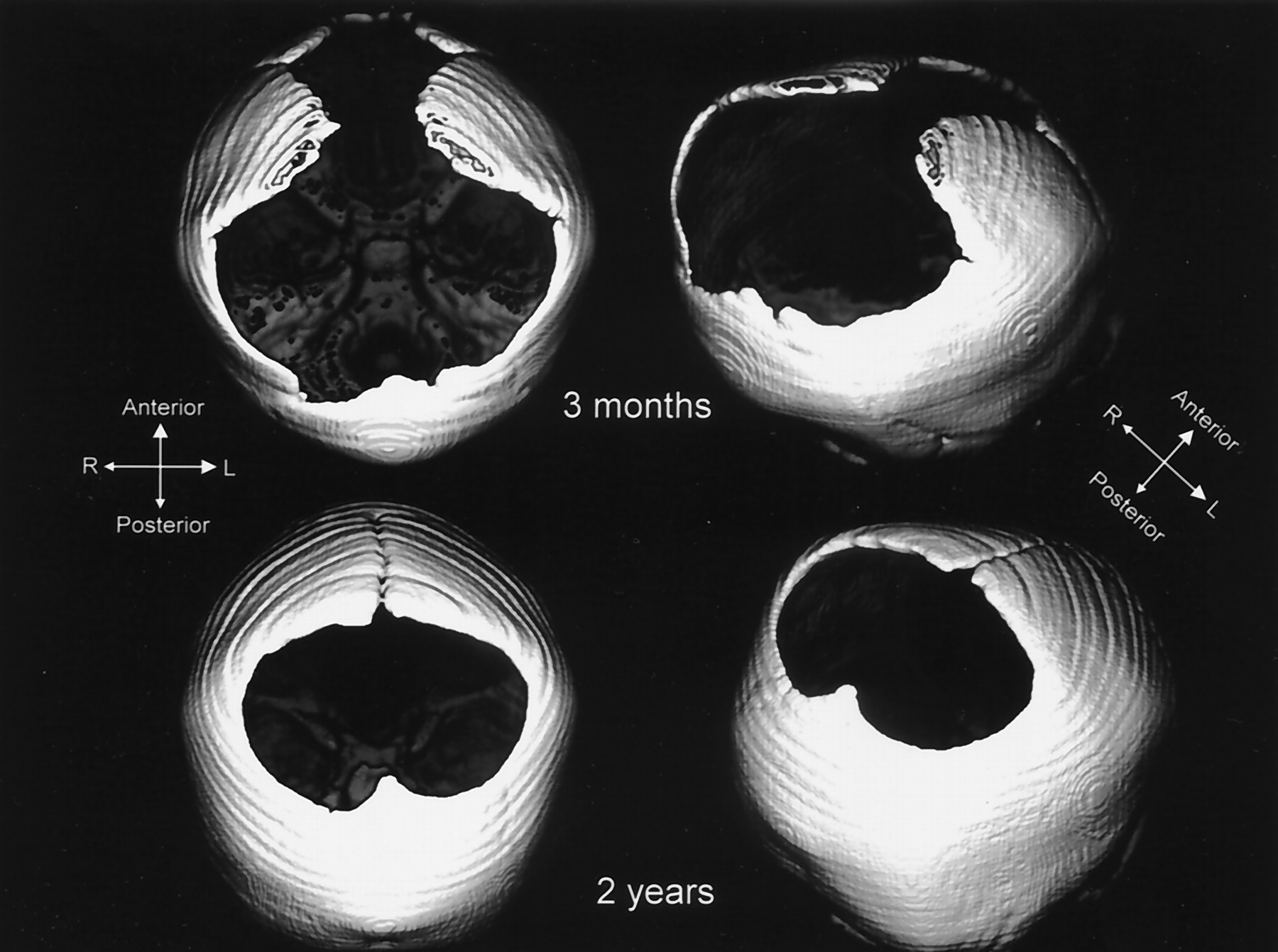
All patients presented with the complete set of abnormalities, although with different levels of severity. A more evident phenotype was found in the newest generation. Moreover, an unambiguous age-related size reduction of the bone alterations was documented in patient III-2 (Fig 3).

CT3D - Patient III-2. Regressive size of the bone defects in time. CT scans at the ages of 3 months and at 2 years of age.
Discussion
This study demonstrated the coexistence of vascular and cortical anomalies in three relatives with PFM2. The presence of very similar anomalies were previously documented by Reedy et al (7) and Sener (8), which corroborates the assumption that these are not incidental findings, but rather were identified because of neuroimaging advances. The study of three generations of one family and the determination of the type of PFM on genetic bases, not previously done by others (7, 8), are major aspects that may provide a better comprehension of the mechanisms that determine the presence and severity of these anomalies in patients with PFM. First, the occurrence of more severe clinical pictures with thrombosis, excessive bleeding during surgical procedures, and epilepsy in some cases of PFM may be explained by the generation-related gravity observed in this study, because the child was the most affected member of this family. In view of the genetic mechanism, recent observations suggested that Alx4 and Msx2 (ALX4 and MSX2 murine homologs) may play similar, but not identical, roles in transcriptional activation, leading to intramembranous ossification of the skull vault (10). To date, there is no evidence of a phenotype-genotype correlation based on published cases (3, 6, 10), although neuroimaging and electroclinical studies of families with MSX2 mutations (PFM1) would be of interest to clarify whether these neuroimaging and neurophysiological abnormalities are more frequent or restricted to PFM2.
It may be postulated that a mesenchymal disturbance could be the primary cause for these tentorial, vascular, and bone abnormalities. It is known that each parietal bone ossifies in a membranous pathway from two ossification centers that make their appearance during the 8th and 9th weeks of fetal life. One of these centers is located adjacent to the upper end of the sigmoid bend of the transverse sinus, close to the tentorial incisura. The tentorium is formed from a coalescence of left and right tentorial membranes that fuse in the midline at 3 months of intrauterine life (8). The falcine sinus, also developed from mesenchyme, is an embryonic vascular channel, originating at the level of the mesencephalic flexure, usually defined as a straight sinus precursor. It tends to close after birth but can be found later in life, and persistence has been associated with an underdeveloped straight sinus and dysplastic tentorium cerebri. Moreover, primitive veins of the posterior fossa have an ascending course in early stages of development. At 12 weeks of gestation, the straight sinus is relatively highly located and, with the increasing volume of the telencephalon, the area where the straight sinus joins the superior sagittal sinus gradually descends. The measurement of the base torcular angle by using fetal MR imaging showed that this angle gradually increases with gestational age (11).
What needs to be addressed is the inter-relationship between these anomalies and the malformation of cortical development in the occipital lobes, adjacent to these structures, as well as the role that ALX4 plays in affecting this chain of events. Our current understanding is malformations of cortical development result from a multifactorial complex pathogenesis with interaction of genetic and environmental factors (12). The inconsistent overlap between cortical abnormalities and the vascular territories of the extrinsic vessels has always argued against a hemodynamic failure. In our case, there is a clear relationship between the abnormal cortex and the vascular abnormality. Thus, an interruption of blood supply during the embryonic period is feasible, although presumably influenced by genetic factors. These theories (vascular and genetic) are not necessarily contradictory, but might be additive. It is possible that the common pathogenetic mechanism is a genetic disorder of perfusion with varying penetration.
Size reduction, illustrated in III-2 and reported extensively in the literature, can be explained by a mechanical or disclosure type defect alone; however, the more prominent intracranial phenotype in the youngest generation, which suggests a generation-modulated expression of intracranial anomaly, is beyond this explanation.
It is known that mutations in mouse Alx-4 affects the craniofacial mesenchyme during embryogenesis and is involved in skull development (10), substantiating the hypothesis that mesenchyme maturity is in some way disturbed when this gene is affected. It is possible that this specific timing and anatomic setting may link the ALX4 homeobox to other clinical situations, especially those involving disorders of the skull vault ossification and vascular and cortical abnormalities. These mechanisms might be further considered in other distinct syndromes in which pathogenic processes remain unclear.
Acknowledgments
We wish to thank Dr. Susan Baser for reviewing this manuscript and making valuable suggestions.
Footnotes
Author’s contributions: Study concept and design (M.V. and K.D.V.); data acquisition (M.V., K.D.V., and S.S.M.S.); data analysis and interpretation (M.V. and C.A.K.); drafting of the manuscript (K.D.V., M.V., C.A.K.); critical revision of the manuscript for important intellectual content (M.V. and C.A.K.); study supervision (M.V. and C.A.K.).
References
- Received December 22, 2003.
- Accepted after revision March 5, 2004.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}