
Abstract
SUMMARY: Hypoxic ischemic encephalopathy may cause early deep white matter abnormalities on MR imaging that usually progress to include gray matter and basal ganglia change. Toxic leukoencephalopathy due to heroin inhalation predominantly causes cerebellar and posterior cerebral radiologic change. Both conditions rarely present clinically and radiologically in a delayed manner with subsequent recovery. We report a case of reversible delayed posthypoxic/toxic leukoencephalopathy with no clinical or radiologic evidence of gray matter insult.
Therapeutic or illicit drug use and hypoxia can cause leukoencephalopathy. The clinical spectrum usually mirrors the severity of white matter change, but a latent period between exposure and the onset of clinical features causing a delayed presentation may occur. Recovery can be complete.
Case Report
A 40-year-old woman was transferred with a 7-day history of neurobehavioral disturbance beginning with confusion and decreased attention and distractability and progressing to include urinary and fecal incontinence. No gait, postural, or mobility abnormalities were reported. MR imaging of the brain at the referring hospital was reported as consistent with acute disseminated encephalomyelitis (Fig 1 A, -B).

A, Initial axial T2-weighted MR image (obtained on day 22 postictus) through the basal ganglia shows diffuse subcortical white matter change with gray matter sparing. B, Initial axial T2-weighted MR image shows extensive bilateral symmetric supraventricular white matter change.
History of back pain had necessitated treatment with morphine, and a pain-related neuropsychologic assessment 24 months before presentation revealed no cognitive deficits. Recent history included an emergency hospital admission 17 days earlier when she was found unconscious of unknown duration with shallow or absent respiratory effort. An apparently inadvertent overdose of morphine (6 60-mg tablets) was subsequently disclosed. Resuscitation for approximately 15 minutes was necessary before intubation and ventilation. Arterial blood gas analysis revealed a profound acidemia (pH = 6.9), presumably respiratory in origin. Intensive care unit admission was complicated by aspiration pneumonia but without further compromise to cerebral oxygenation. Recovery was reported as complete 7 days after admission, allowing discharge. The first week of recuperation was uneventful, but behavioral problems manifested 10 days later.
On neurologic presentation, the patient was alert and independently mobile. She was aware of and admitted to all events preceding hospitalization but had nonspecific headache and visual disturbance. On observation, she was restless, fidgety, and distractible. Findings of a general medical examination were normal. Neurologic examination findings demonstrated no cranial nerve or limb abnormalities. Bedside cognitive assessment revealed echolalia and perseveration, with mimicry of actions and tasks observed, but some questions were answered appropriately. Personal care was poor, with prompting required for basic toileting needs. Addenbrooke’s Cognitive Examination (ACE)1 yielded a score of 40/100, consistent with severe global cognitive impairment. The deficits were most profound in memory and verbal fluency.
The results of hematologic, biochemical and immunologic profiling, toxicology screening, carboxyhemoglobin estimation, and assessment of lysosomal white cell enzyme activity were all unremarkable. Electroencephalography confirmed a significant encephalopathic process with excess delta activity. Lumbar puncture yielded clear CSF with normal parameters. The most profound abnormality at this stage was radiologic (Fig 2A–D).
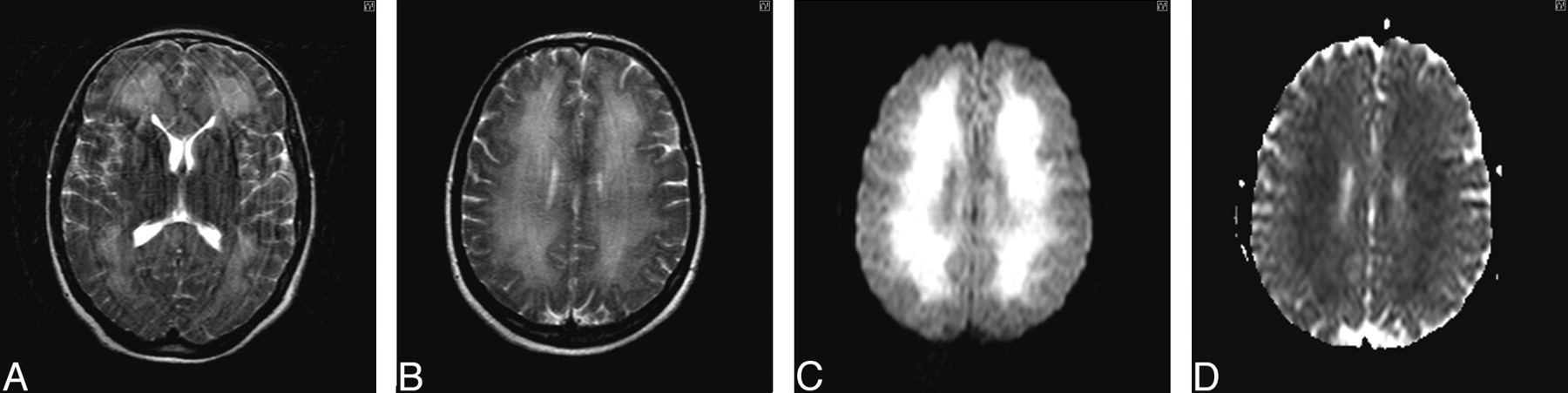
A and B, Axial T2-weighted MR image of brain (obtained on day 24 postinsult) demonstrates extensive bilateral symmetric white matter hyperintensity extending subcortically, with sparing of the basal ganglia deep gray matter and cortical structures. C, Diffusion-weighted image (DWI) obtained 24 days after initial insult shows leukoencephalopathy, with high signal intensity on isotropic imaging. D, Apparent diffusion coefficient (ADC) mapping confirms restricted diffusion of the cerebral white matter.
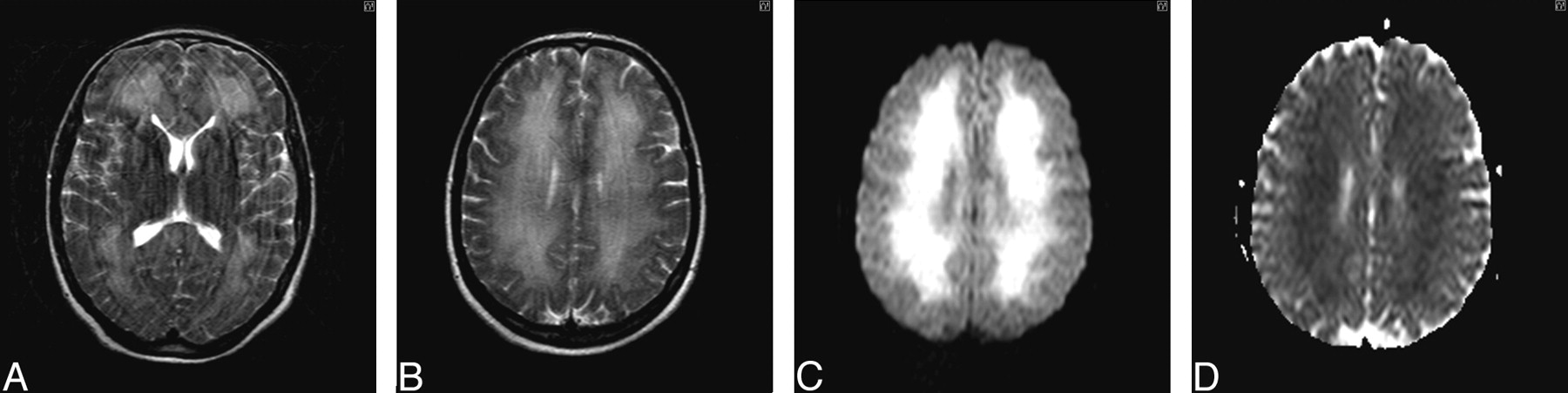
Clinical improvement occurred with supportive care only. The results of repeat ACE at day 34 were 72/100 with persistent deficits in orientation and memory. Further improvement was observed during the next 9 months. Repeat imaging was performed 6 months after the ictus (Fig 3A–D). Repeat ACE at 9 months confirmed cognitive recovery with a score of 97/100. The patient had returned to work.

A and B, T2-weighted axial MR image of brain obtained 6 months after initial presentation shows improvement in white matter hyperintensities without volume loss. C, DWI obtained 6 months after initial presentation demonstrates a resolving leukoencephalopathy. D, ADC mapping confirms resolution of the previously restricted diffusion profile.
Discussion
Acute hypoxic encephalopathy typically causes radiologic and pathologic insult to gray matter, which is thought more vulnerable to diffuse ischemic injury than neighboring white matter. Chalela et al2 reported that early MR imaging in these cases, performed within 6 days of symptom onset, has demonstrated periventricular white matter change with restricted diffusion, suggestive of an acute myelinopathy with cytotoxic edema. Almost half of the patients in their report had concomitant radiologic gray matter change. The etiology of hypoxia varied from cardiorespiratory compromise to unspecified toxic causes. None of their patients made a clinical recovery, and long-term radiologic data were not reported.
Toxic leukoencephalopathy has been associated with a variety of “leukotoxins,” including carbon monoxide, solvents, chemotherapeutic agents, cranial irradiation, and drugs of abuse (alcohol, cocaine, and heroin). Clinical features usually mirror radiologic and pathologic damage, with presentation most frequently due to altered mental status. Diagnosis should not be made without corroborative evidence from imaging, with T2-weighted MR imaging the technique of choice.3 Fatal acute toxic leukoencephalopathy, associated with diffuse white matter abnormalities on MR imaging, has been histologically correlated with deep white matter vacuolar degeneration, with adjacent axonal injury in normal-appearing white matter.4
Heroin vapor inhalation–induced leukoencephalopathy typically causes symmetric T2-weighted MR imaging hyperintensity, affecting the cerebellum and pyramidal projections, particularly the posterior limb of the internal capsule.5 Gacouin et al reported that radiologic change is crucial for diagnosis, particularly because the history of the patient is often unreliable and complicated by hypoxia, with physical findings nonspecific. Acute toxic leukoencephalopathy caused by heroin inhalation can be reversible, with radiologic resolution reported after 7 months. Clinical recovery was not complete with persistent ataxia despite impressive cognitive improvement. The use of antioxidants, such as ubiquinone (Coenzyme Q), may aid resolution of the clinical and radiologic syndrome6.
Delayed posthypoxic leukoencephalopathy has been documented most extensively in patients with carbon monoxide poisoning. Choi7 reported that only 2.75% of patients exposed to carbon monoxide presented in this way. The mean delay in symptom onset was 2.4 days but ranged from 2–40 days. Radiologic abnormalities were reported on CT imaging, but 54% of findings were documented as normal. The abnormal scans predominantly demonstrated basal ganglia hypointensities, with 2 reports of decreased attenuation in the cerebral white matter. Outcome was variable, but complete recovery was documented in 75% of patients within 1 year.
Delayed toxic leukoencephalopathy due to heroin may have a poor outcome. Rizzuto et al8 reported on a patient with coma induced by intravenous heroin overdose who had a fatal neurologic deterioration 3 weeks after initial insult, described as consistent with a delayed encephalopathy. At autopsy, this patient had evidence of diffuse gliosis with spongiform change, without the hypoxic necrotic lesions of the basal ganglia previously associated with delayed posthypoxic encephalopathy.
Abnormalities in arylsulfatase enzyme activity, leading to either complete or incomplete deficiencies, may predispose the development of posthypoxic leukoencephalopathy. Gottfried et al9 described a 36-year-old man with arylsulfatase A deficiency who presented after oral morphine overdose. An initial full recovery was followed by behavioral changes, myoclonus, and incontinence on day 24. MR imaging of the brain demonstrated extensive bilateral white matter hyperintensity on T2-weighted sequences. Repeat scanning after further neurologic deterioration (spastic quadriparesis, emergence of primitive reflexes, dysautonomia, and stupor) on day 34 demonstrated new bilateral hyperintensity in the globus pallidus. Recovery was significant but incomplete, with persistent amnesia, spasticity, and care dependence reported after 6 months. Follow-up neuroradiologic assessment was not documented.
By contrast, Lee et al10 described a 71-year-old patient who presented after accidental benzodiazepine overdose, loss of consciousness, and possible aspiration pneumonia. Despite initial complete recovery after 3 days, re-presentation with neurobehavioral abnormalities and incontinence occurred 14 days later. On this occasion, findings of MR imaging of the brain were reported as normal. Clinical deterioration with severe cognitive impairment and an extrapyramidal gait disorder ensued. On day 24, MR imaging demonstrated diffuse periventricular white matter change thought consistent with delayed posthypoxic leukoencephalopathy. An apparent full cognitive and physical recovery was described 6 months after the initial insult. Repeat imaging was not reported.
Delayed hypoxic-ischemic leukoencephalopathy may occur because the widely spaced linear arterioles and lack of anastomoses of the deep white matter render it vulnerable to hypoxic ischemia.11 Metabolic deficits and/or adulterants in drugs of abuse have been proposed to precipitate this presentation, but the etiology remains poorly understood. The delayed but purely neurobehavioral presentation, despite extensive radiologic leukoencephalopathy caused by hypoxia due to oral morphine overdose, in the absence of white cell enzyme abnormalities, makes our case noteworthy. Although this condition is potentially fatal, spontaneous clinical and neuroradiologic recovery may be complete within a relatively short time.
References
- Received September 1, 2005.
- Accepted after revision September 26, 2005.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Toxic leukoencephalopathy versus delayed post-hypoxic leukoencephalopathy after oral morphine sulphate overdose
- Outcomes associated with hospital admissions for accidental opioid overdose in British Columbia: a retrospective cohort study
- Dynamic MR Imaging Patterns of Cerebral Fat Embolism: A Systematic Review with Illustrative Cases