
Abstract
SUMMARY: We describe a case of endolymphatic sac tumor with drop metastasis to the spine. Our review of the literature showed that this is only the 2nd reported case of such an occurrence.
Endolymphatic sac tumors (ELSTs) are rare neuroectodermal neoplasms of the petrous temporal bone. Patients present with symptoms including insidious onset of neurosensory hearing loss, tinnitus, and facial nerve palsy. These tumors can be confused with other tumors of the petrous temporal bone or posterior fossa on the basis of both the radiologic and histopathologic characteristics. Although ELSTs are known to be locally aggressive, they very rarely metastasize. We describe a case of ELST with metastases to the spine. To our knowledge, this is only the 2nd case of drop metastases reported in the literature.1
Case Report
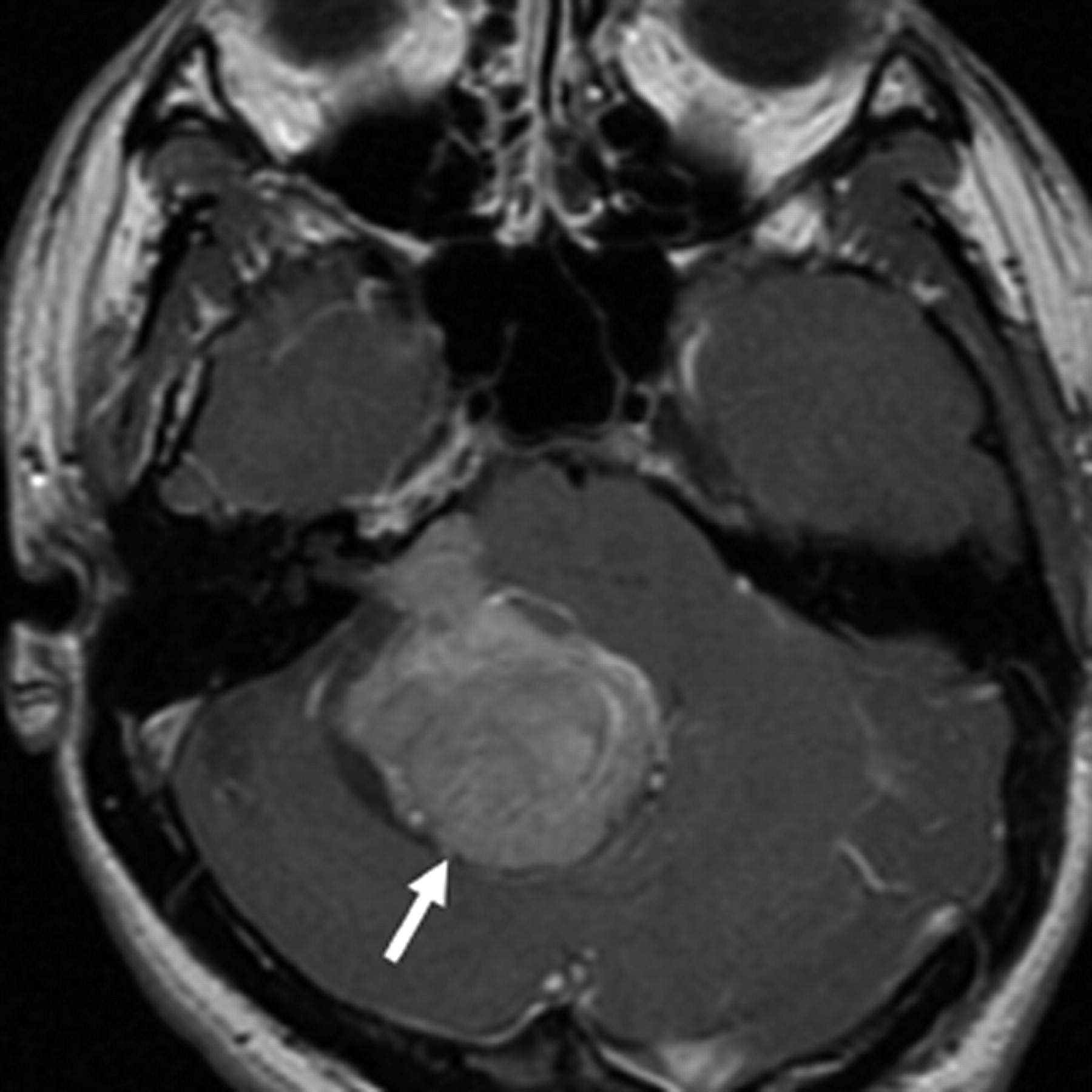
A 27-year-old man with a history of endolymphatic sac tumor was referred for MR imaging of the spine because of saddle numbness. He first presented 5 years earlier with a 6-month history of headaches, nausea, vomiting, and diplopia. CT revealed a right posterior fossa mass with internal calcifications that was causing obstructive hydrocephalus (Fig 1). MR imaging showed an extra-axial mass in the right cerebellopontine angle. The lesion was predominantly solid with cystic components. It was hypointense on T1- and isointense on T2-weighted sequences, with homogeneous enhancement of the solid components (Fig 2 and 3). Imaging differential diagnoses included atypical acoustic neuroma, meningioma, and schwannoma of the adjacent cranial nerves.

Axial CT of the brain shows a partly calcified posterior fossa mass (white arrow), which is compressing the 4th ventricle and causing hydrocephalus. Note the dilated temporal horns of the lateral ventricles (white arrowheads).

Axial T2-weighted image (TR/TE, 4400/90) shows an isointense mass (white arrow) in the right posterior fossa. Note that there is no extension into the internal auditory canal (white arrowhead).

Axial contrast-enhanced T1-weighted image (TR/TE, 816/20) reveals homogeneous enhancement in the right posterior fossa mass (white arrow).
The tumor was resected via a retrosigmoid approach. The vascular mass arose from the posteroinferior aspect of the right petrous ridge without involvement of the underlying bone.
Histopathologic examination confirmed ELST. It showed distinctive immunohistochemical reactivity for periodic-acid-Schiff (PAS), low-molecular-weight cytokeratin, S100, synaptophysin, and neuron-specific enolase (NSE).
Clinical examination revealed no suspicion of von Hippel-Lindau (VHL) disease. Findings of CT examinations of the chest, abdomen, and pelvis were normal.
The tumor recurred 2½ years later, presenting as a small dural-based nodule along the right lateral aspect of the foramen magnum. Adjuvant radiation therapy (50 Gy in 25 fractions) to the primary and recurrent sites was given following excision of the recurrent tumor.
More recently, the patient reported symptoms of saddle anesthesia. MR imaging demonstrated multiple enhancing intradural extramedullary nodules within the lower thoracic and lumbosacral spine (Fig 4). The appearance was consistent with drop metastases. There was no known history of any other malignancies. The clinicians decided not to perform CSF sampling and opted to treat the patient with palliative radiation therapy (3000 cGy in 10 fractions).

Sagittal T2-weighted (TR/TE, 5350/112) (A) and postgadolinium-enhanced T1-weighted (TR/TE 666/8) (B) images show multiple enhancing intradural extramedullary masses (white arrows) in the central canal of the lumbar spine.
Discussion
Adenomatous tumors of the temporal bone have been described for more than a century. More recently, these lesions have been further classified as nonpapillary middle ear adenomas and the more aggressive papillary middle ear tumors.2 Hassard et al3 were the first to report an adenoma arising from the endolymphatic sac. Heffner4 suggested that the more aggressive papillary tumors be classified as low-grade papillary adenocarcinoma of the temporal bone arising from the endolymphatic sac. The term “endolymphatic sac tumor” was first proposed by Li et al in 1993.5
Most ELSTs occur sporadically. Patients with VHL disease have a higher incidence of ELSTs. VHL gene mutations have been demonstrated in ELST specimens of both sporadic and VHL-associated ELSTs.6,7 Endolymphatic sac and duct epithelium of patients with VHL disease are found to have microscopic abnormalities similar to those of ELSTs, even when the patients are clinically and radiologically healthy.7 Bilateral ELSTs are more commonly reported in patients with VHL disease (28% versus 1%). Patients with ELSTs and VHL are twice as likely to be female as those with sporadic ELSTs.8
ELSTs arise from the endolymphatic duct or sac, in the posterior aspect of the petrous temporal bone.5,7,9 Involvement of the petrous apex with extension into the cerebellopontine angle and posterior cranial fossa is common. This accounts for the development of sensorineural hearing loss. Other complaints include tinnitus, facial weakness/paralysis, ataxia, and disequilibrium. Many patients have been misdiagnosed initially as having Meniere disease.
Imaging typically reveals a retrolabyrinthine mass. CT usually demonstrates a lytic lesion within the petrous temporal bone with irregular bone margins and intratumoral bone spicules. There is typically avid enhancement and central areas of cystic change or necrosis. MR imaging reveals a soft-tissue mass with heterogeneous enhancement. High-intensity foci may represent methemoglobin or proteinaceous collections, whereas low-intensity areas represent hemosiderin or calcifications. ELSTs are vascular lesions and are commonly supplied by branches of the external carotid artery (ascending pharyngeal and stylomastoid arteries). As the tumor grows, it may draw blood supply from the internal carotid artery or the posterior circulation.10
There are 2 broad histopathologic categories: a follicular form containing colloid-filled cysts and a sparse stroma, which can mimic thyroid tissues, and a more solid and papillary form, in which cells with clear cytoplasm and central nuclei are prominent. These resemble plant cells and can look like renal cell carcinoma.9
ELSTs have commonly been misdiagnosed radiologically. Differential diagnoses include paraganglioma, metastases, meningioma, chondrosarcoma, hemangiopericytoma, and plasmacytoma. Histopathologically, it can be difficult to differentiate ELST from metastatic thyroid carcinoma, renal carcinoma, paraganglioma, ceruminal gland tumor, salivary choristoma, and choroid plexus papilloma.
Immunohistochemical staining has been helpful in the diagnosis of ELSTs. They are noted to stain positive for PAS, cytokeratin, S100, vimentin, NSE, and epithelial membrane antigen.9 A high MIB-1 index (Ki-67 antigen-expressing fraction) may indicate a more aggressive tumor in an otherwise low-grade malignancy.11
Metastases from ELST have rarely been reported in the literature. Bambakidis et al1 reported a patient presenting with a history of radicular symptoms many years after prior surgical resections and stereotactic radiosurgery. Imaging revealed an enhancing intradural mass in the midlumbar region. Surgical resection revealed metastatic ELST.
ELSTs are rare, and metastases from ELSTs are even more uncommon. Thus, routine screening for spinal metastasis is unlikely to be cost-effective. However, spinal imaging should be considered in patients with specific complaints such as back pain, cauda equina, and radicular symptoms.
Conclusion
We described a case of ELST with drop metastases to the spine. The previously held opinion that ELSTs do not metastasize is now questionable because this is the 2nd reported case of drop metastasis in the spine.
References
- Received March 5, 2006.
- Accepted after revision May 15, 2006.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}