
Abstract
BACKGROUND AND PURPOSE: Hereditary spastic paraplegia (HSP) is a disorder characterized by degeneration of the corticospinal tracts and posterior column of the spinal cord. Previously described radiologic findings included nonspecific brain abnormalities such as brain atrophy and white matter lesions, as well as atrophy of the spinal cord. In our study, we aimed to better characterize brain and spine MR imaging findings in a series of patients with HSP.
MATERIALS AND METHODS: Nine patients from 4 different Lebanese families with the autosomal recessive form of HSP were included in the study. All patients underwent brain and whole-spine MR imaging. We assessed the presence of white matter abnormalities mainly along the corticospinal tracts, brain atrophy, thinning of the corpus callosum, and the presence of spinal cord atrophy or abnormal signal intensity.
RESULTS: Imaging revealed mild brain atrophy (44%), atrophy of the corpus callosum (55%), white matter lesions (67%), abnormal T2 high signal intensity in the posterior limb of the internal capsule (55%), and mild spinal cord atrophy (33%).
CONCLUSIONS: The MR imaging findings of HSP are nonspecific and variable; however, the most prominent features include atrophy of the corpus callosum, T2 signal intensity in the posterior limb of the internal capsule, and spinal cord atrophy.
Hereditary spastic paraplegia (HSP) is a heterogeneous group of hereditary disorders first described by Strumpell in 1880, characterized by degeneration of the corticospinal tracts and posterior column of the spinal cord. They are usually divided into 2 groups: a “pure” form and a “complicated” form. The latter comprises, in addition to spastic paraplegia, cerebellar symptoms, amyotrophy of the upper limbs, extrapyramidal signs, ophthalmoplegia, optic neuropathy, retinal pigmentary changes, skin disorders, mental retardation, and peripheral neuropathy. Dominant, recessive, and X-linked patterns of inheritance have been described. Approximately 70% of cases follow a dominant pattern of inheritance, and 20% show a recessive pattern.1,2 Recent genetic studies have identified multiple loci on different chromosomes responsible for autosomal recessive, autosomal dominant, or X-linked forms of HSP and some of the proteins encoded by those genes.3–5 The radiologic findings of HSP are nonspecific, including mild-to-moderate brain atrophy, thinning of the corpus callosum, nonspecific white matter lesions in the cerebral hemispheres, abnormal T2 high signal intensity in the posterior limb of the internal capsules, and atrophy of the spinal cord.3–10
Pathologic studies have shown that the corticospinal tracts, which route through the posterior limb of the internal capsules on their way to the brain stem and spinal cord, are the main tracts involved in HSP. Recently, in brain MR imaging of 2 unrelated patients with autosomal recessive HSP, we found high signal intensities in the posterior limb of the internal capsules bilaterally in the anatomic location of the corticospinal tracts. On the basis of these MR imaging findings, we decided to characterize the brain and spine MR imaging findings in a series of patients with HSP.
Materials and Methods
Nine patients from 4 different Lebanese families with the autosomal recessive form of HSP were included in the study. All of the patients were born at full term, had no reported history of perinatal complications, and achieved normal developmental milestones. The study was approved by the institutional review board of the American University of Beirut.
Family I comprised patients 1–4; 3 men and 1 woman; 30, 28, 26, and 22 years of age, respectively. The parents were first-degree cousins and were clinically unaffected. Their oldest daughter, 31 years of age, was clinically unaffected. Family II comprised 19-year-old twin girls (patients 5 and 6), who were the product of a consanguineous marriage, with 2 unaffected brothers. Family III comprised patients 7 and 8, a 26-year-old man and a 16-year-old girl. Their parents were first-degree relatives, and they had 2 brothers and 2 sisters, who were reportedly healthy. Family IV comprised patient 9, a 42-year-old man, who had a brother and a sister who were reportedly healthy. His parents were first-degree paternal cousins. He had 3 children, all of whom were reportedly healthy but were still younger than 20 years of age.
The diagnosis of HSP was based on the criteria adapted from Harding,11 and an autosomal recessive pattern of inheritance was diagnosed on the basis of the criteria adapted from Coutinho et al.12 The differential diagnosis of HSP includes structural spinal cord abnormalities, degenerative diseases such as amyotrophic lateral sclerosis, leukodystrophies, and metabolic and infectious disorders. Work-up, including blood studies, neurography and electromyography, and imaging studies in addition to the clinical findings and family history, was performed to rule out these entities. Genetic testing for these patients was not performed. All patients underwent brain and whole-spine MR imaging by using a 1.5T magnet. The brain was evaluated with sagittal spin-echo (SE) T1-weighted images (TE, 13 ms; TR, 400 ms), axial and coronal fluid-attenuated inversion recovery (FLAIR; TE, 150 ms; TR, 6000 ms; TI, 2000 ms), and axial T2 gradient and SE-weighted images (TE, 120 ms; TR, 3052 ms). Sagittal and axial turbo SE T2-weighted images were obtained at the level of the spinal cord (TR, 3500 ms; TE, 120/140 ms), with the axial images obtained parallel to the intervertebral disk space. The MR images were transferred to a workstation for further analysis and measurement of the diameter of the spinal cord. All MR images were read and interpreted by 2 neuroradiologists who were blinded to the clinical examination and laboratory studies. They assessed the following MR imaging abnormalities:
Brain white matter abnormalities on T2-weighted and FLAIR images, mainly along the course of the corticospinal tracts. White matter abnormalities were graded as absent, mild, moderate, or severe.
Brain or cerebellar atrophy was divided into 3 grades: mild, moderate, or severe.
Thinning of the corpus callosum was classified as normal and mildly, moderately, or severely atrophic.
Spinal cord abnormal signal intensity or atrophy.
The severity of the findings was graded subjectively by the 2 neuroradiologists because, to our knowledge, there is no international standard rating scale for brain atrophy and white matter lesions.
Other pathologic abnormalities were also noted.
Results
Clinical Findings
A total of 9 patients (5 male and 4 female) from 4 different families were included in the study. All of them fulfilled the criteria for the diagnosis of HSP. The age onset of symptoms ranged from 18 months to 17 years (mean age of onset, 9 years). All of them started with symptoms consisting of gait difficulties and weakness in the lower extremities, and their neurologic examination showed an upper motor neuron syndrome predominantly in the lower extremities with weakness, spasticity and hyperreflexia, and bilateral extensor plantar response. Two patients (family III) had the pure form of HSP. Seven patients (families I, II, and IV) had additional symptoms and were included in the category of complicated HSP. Four patients (family I) had dementia, which resulted in a very poor school performance. Five patients (2 from family I, 2 from family II, and 1 from family IV) had a peripheral neuropathy diagnosed by neurography and electromyography consisting of axonal motor neuropathy. Cerebellar signs were present in 4 patients (family I) and consisted of a dysarthric cerebellar speech, dysmetria, and abnormal rapid alternating movements in the upper extremities. Vibratory sense was affected in all patients and ranged from absent to severely abnormal (families I and IV) and from mild to moderately abnormal (families I and III). Two patients (family II) had distal amyotrophy. Three patients had musculoskeletal deformities (2 patients from family I had scoliosis and 1 patient from family IV had pes cavus and hammertoes). One patient (family I) had optic atrophy. None of the patients exhibited extrapyramidal symptoms or signs.
Imaging Findings
All of the patients underwent brain and whole spine MR imaging. The age of patients at imaging studies varied between 16 and 42 years.
The T2 high-signal-intensity lesions in the white matter were present in 6 of 9 patients (67%), all members of families I and III. Those white matter lesions were mild and located in the periventricular white matter and centrum semiovale. In family I, the white matter lesions were more prominent in older patients (Fig 1A), in contrast to family III, where the white matter lesions were more prominent in the younger patients. T2 abnormal high signal intensity along the posterior limb of the internal capsule bilaterally was described in 5 patients (55%) (All patients in family I and the older patient in family III) (Fig 2A, -B).

Axial FLAIR (A) and sagittal T1-weighted images (B) of patient 1. A, Note T2 high signal intensity in the periventricular white matter and corona radiata. B, This patient also has a very thin corpus callosum as shown on the sagittal image.
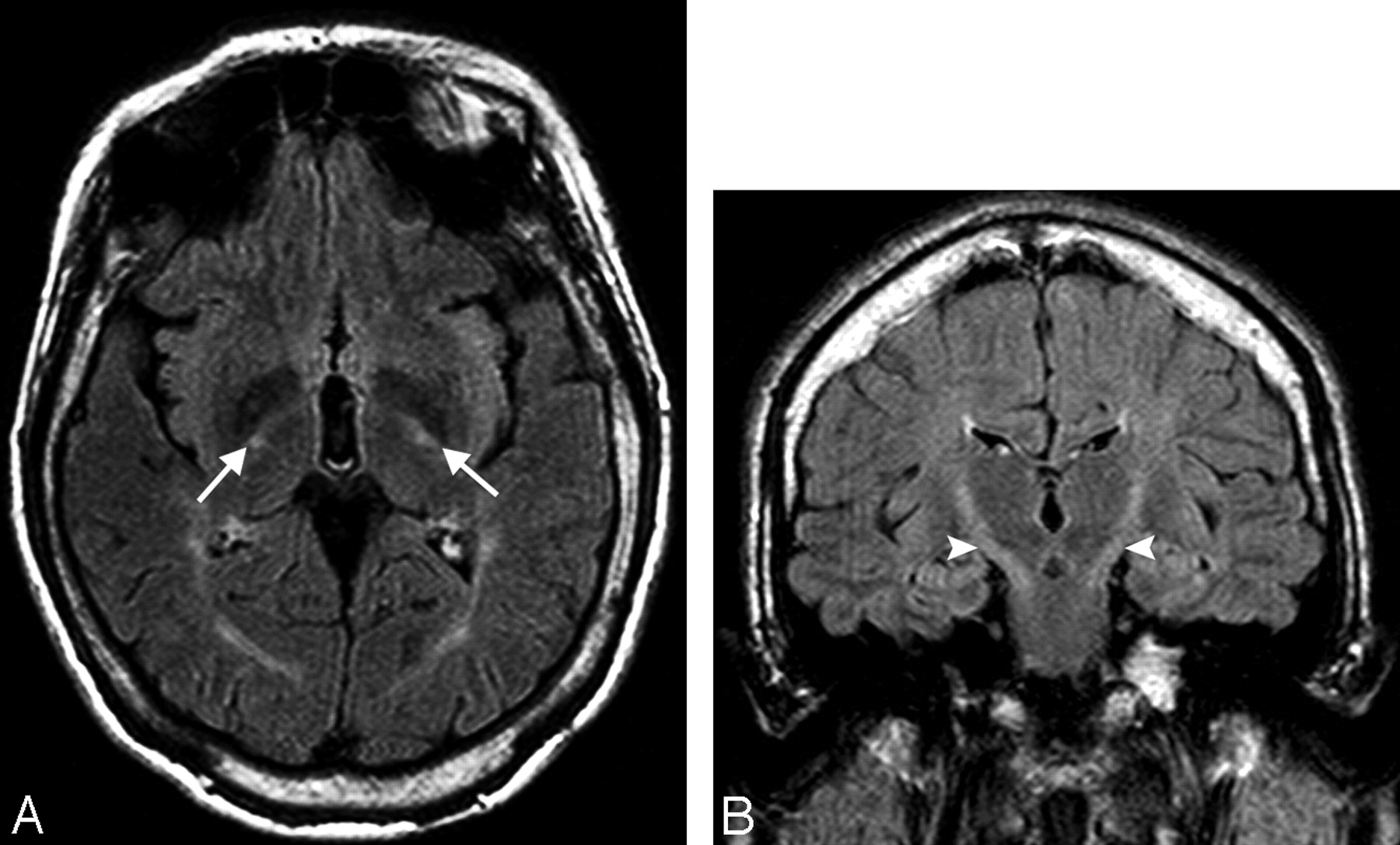
Axial and coronal FLAIR images of patient 4 show high signal intensity in the posterior limb of both internal capsules (white arrows, A) and along the corticospinal tracts (white arrowheads, B).
Mild brain atrophy was seen in patients 1, 2, 7, and 8. Patient 7 also had mild cerebellar atrophy. The thinning of the corpus callosum was seen in all 4 patients belonging to family I (44% of cases) (Fig 1B). It was small, hypoplastic, and severely atrophic. In the spinal cord, patient 1 had mild atrophy of the cervical spinal cord compared with the rest of his family members. In patients 5 and 6 of family II, there was T2 high signal intensity along the posterior aspect of the cervical and, to a lesser extent, the dorsal spinal cord, which may represent either a posteriorly displaced central ependymal canal secondary to posterior column atrophy and degeneration or high signal intensity along the dorsal column of the cord (Fig 3A, -B).

Sagittal and axial T2-weighted images of the cervical spinal cord of patient 5. There is a linear T2 hyperintense signal intensity in the posterior aspect of the spinal cord (white arrows, A), which represents, on the axial image (white arrow, B), an enlarged and posteriorly displaced ependymal canal.
Discussion
Our patients satisfied the clinical criteria for diagnosis of HSP as proposed by Harding in 1984.11 Most had an autosomal recessive pattern of inheritance, according to the criteria proposed by Coutinho et al.12 Seven of 9 patients belonged clinically to the class of complicated HSP because they had, in addition to spastic paraplegia, other findings, including dementia, peripheral neuropathy, cerebellar signs, ophthalmologic findings, and distal amyotrophy. Neuropathologic studies looking at the brain and spinal cord of patients with HSP revealed neurodegeneration and gliosis affecting a large portion of the central nervous system: the corticospinal tracts, thalamus, cerebral white matter, substantia nigra, cerebellum, anterior and lateral corticospinal tracts, spinocerebellar tracts, and posterior columns, mainly the fasciculus gracilis of the spinal cord. HSP is a retrograde axonal degeneration or a “dying back” process, starting at the distal portion of the corticospinal tracts, the fasciculus gracilis, and the spinocerebellar tracts and progressing cranially toward the cell body. The rest of the neurons showed an excessive amount of lipofuscin and eosinophilic intracytoplasmic granules, reflecting accelerated brain aging.9,13–15
Radiologically, brain and spinal MR imaging is usually used to rule out other diseases included in the differential diagnosis of HSP. Whereas findings of brain and spinal MR imaging may appear normal in some patients as in our patient 9, there is a spectrum of MR imaging findings seen in patients with HSP, including mild brain and spinal atrophy, high T2 signal intensity in the posterior limb of the internal capsule, unspecific white matter lesions, thinning of the corpus callosum,3–10 and brain and cerebellar atrophy predominantly in the motor areas and pericentral gyri.5 A quantitative 3D MR imaging study of the brain volume demonstrated marked brain atrophy associated with HSP compared with age-matched controls, and it was more severe and involved both gray and white matter16 in the complicated forms of HSP. In our study, 4 patients had mild cerebral atrophy and just 1 had mild cerebellar atrophy (patient 7).
Another important finding is the abnormal T2 high signal intensity in the posterior limb of the internal capsule at the site of the descending fibers of the corticospinal and corticobulbar pathways, extending from the corona radiata through the posterior arm of the internal capsule to the brain stem.5,15,17 This sign was seen in 5 of our patients (55%) (Fig 2A, -B). In family III, it was present in the older patient but absent in the younger one.
Nonspecific white matter hyperintense lesions in the periventricular white matter and centrum semiovale mainly in the temporoparietal areas were also described in the literature. They are prominent in older patients, suggesting ongoing axonal degeneration and an advanced state of disease.15,18 In our study, these white matter lesions were mild, observed in (67%) of patients, and were slightly more prominent in the older than younger patients within the same family (family I) (Fig 1A). In family III, the white matter lesions were more prominent in the younger member. This observation may reflect both gene penetrance and age-related accumulation of lesions. These nonspecific white matter lesions should be carefully interpreted because some studies demonstrate no significant difference between patients with HSP and a healthy population of the same age.19
Another reported radiologic finding in HSP is a small corpus callosum and “corpus callosum index,” which has been described in the complicated autosomal recessive forms linked to chromosome 15q13–q15, mainly in Japanese patients7–9,20–22; more recently, a thin corpus callosum has been described in patients with autosomal dominant HSP related to the spastin gene mutation.23,24 The etiology of a thin corpus callosum is still debated concerning whether it is secondary to congenital hypoplasia or progressive atrophy. Some authors consider it hypoplasia occurring early in the course of the disease, and the thickness of the corpus callosum remains stable with time in the follow-up examinations.8,20,25,26 However, Kuru et al9 and Casali et al27 reported cases with progressive thinning of the corpus callosum and suggested progressive atrophy of the corpus callosum, a neurodegenerative process secondary to severe gliosis in the cerebral white matter.
In our study, the atrophy of the corpus callosum was present in 44% of patients. It was predominant in the posterior aspect of the corpus callosum (Fig 1B) and was associated with white matter changes in the posterior corona radiata and peritrigonal areas, in contrast to the cases described by Teive et al20 and Somasundaram et al,28 where the thinning was mainly along the anterior aspect of the corpus callosum and associated with frontal lobe atrophy. Nevertheless, in all our patients, there was a relationship between the location and severity of white matter changes and the atrophy of the corpus callosum, suggestive of an atrophic process of the corpus callosum.
The new MR imaging techniques like proton MR spectroscopy, diffusion tensor imaging (DTI), single-photon emission CT (SPECT), and fluorodeoxyglucose−positron-emission tomography (FDG-PET) have rarely been reported in patients with HSP.29 Single-voxel proton MR spectroscopy revealed a reduced concentrations of N-acetylaspartate (NAA), creatine (Cr), and choline (Cho) and elevated levels of myo-inositol. These abnormalities showed progression during a period of 5 years. NAA is a neuronal marker; its reduction indicates loss of neuroaxonal tissue. Cr is an energy metabolism marker and is reduced in areas of decreased cellular attenuation. Cho is a marker of membrane synthesis and degradation and is a marker of myelin turnover; reduced concentration is seen in hypomyelination and dysmyelination. Myo-inositol is a marker of astrocytic proliferation.30 Those metabolic changes are consistent with the progressive neuroaxonal loss and astrocytic proliferation known to occur in HSP.29 DTI showed increased mean diffusivity and reduced fractional anisotropy in the periventricular white matter, compatible with damaged myelinated axons.
SPECT and FDG-PET demonstrated decreased blood flow and hypometabolism in the thalamus and in the frontal, temporal, and parietal cortex.31 A longitudinal clinical and neuroradiologic follow-up demonstrated further decrease of relative cerebral blood flow in the thalamus or cerebral cortex suggestive of progressive thalamic and cerebral cortical involvement.21,32
At the level of the spinal cord, studies by Sperfeld et al33,34 showed significant atrophy of the upper spinal cord in patients with HSP compared with the controls, both at the cervical and thoracic levels. A study by Krabbe et al7 demonstrated significant decrease in the anteroposterior diameter of the spinal cord at the T3 and T9 levels in patients with autosomal dominant pure HSP compared with controls, which corresponded neuropathologically to degeneration of the lateral corticospinal tracts, uncrossed pyramidal tracts, and fasciculus gracilis (posterior columns of the spinal cord) from the lumbar level up to the upper cervical level. Another study by Hedera et al4 showed decrease in the cross-sectional area of the spinal cord at the C2 and T9 levels compared with healthy controls. The cord atrophy was more prominent in patients with spastin gene mutations SPG6 and SPG8 HSP than in subjects with SPG3 and SPG4 types.
The previous MR imaging studies could not analyze separately the size of the different anterior (descending) and posterior (ascending) spinal cord tracts responsible for the great part of the atrophy of the spinal cord. However, they did speculate that the atrophy is predominantly secondary to the degeneration of the descending pyramidal tract, resulting in marked atrophy at the thoracic and not at the cervical level.4 Mild atrophy of the cervical and thoracic spinal cord (not quantified) was also reported by Durr et al,3 Lesca et al, 5 Nicolau et al, 10 and Nielsen et al.35 In our study, patient 1 had mild atrophy of the cervical spinal cord and 2 patients of family II presented an unusual finding, namely the round T2 high signal intensity in the posterior aspect of the spinal cord representing either an enlarged and posteriorly displaced ependymal canal or a direct high signal intensity along the dorsal columns of the cord (Fig 3A, -B); in both cases, it reflects the neuropathologic finding of degeneration and atrophy of the dorsal columns of the spinal cord. Although this finding is not very conclusive in confirming the diagnosis of HSP, it may be useful in the evaluation of dorsal column degeneration.36
Conclusions
HSP is a group of neurodegenerative disorders, genetically heterogeneous and progressive with time, characterized by multisystem degeneration. The MR imaging findings are nonspecific; however, the most known characteristic features include atrophy of the corpus callosum, T2 high signal intensity in the posterior limb of the internal capsule, and spinal cord atrophy. These neuroradiologic findings are not constant; they vary among the different phenotypes of HSP and according to the stage of the disease. Furthermore, in the same family with supposedly the same genetic profile, the degree and location of axonal degeneration may differ depending on the gene penetrance.
Footnotes
This work was supported by the University Board for Research to perform diagnostic procedures.
References
- Received October 16, 2008.
- Accepted after revision December 5, 2008.
- Copyright © American Society of Neuroradiology





{kind=link}
{kind=link}
{kind=link}