
Abstract
BACKGROUND AND PURPOSE: Rabbit aneurysm models are used for the testing of embolization devices and elucidating the mechanisms of human intracranial aneurysm growth and healing. We used RNA-sequencing technology to identify genes relevant to induced rabbit aneurysm biology and to identify genes and pathways of potential clinical interest. This process included sequencing microRNAs, which are important regulatory noncoding RNAs.
MATERIALS AND METHODS: Elastase-induced saccular aneurysms were created at the origin of the right common carotid artery in 6 rabbits. Messenger RNA and microRNA were isolated from the aneurysm and from the control left common carotid artery at 12 weeks and processed by using RNA-sequencing technology. The results from RNA sequencing were analyzed by using the Ingenuity Pathway Analysis tool.
RESULTS: A total of 9396 genes were analyzed by using RNA sequencing, 648 (6.9%) of which were found to be significantly differentially expressed between the aneurysms and control tissues (P < .05; false-discovery rate, <0.01; fold change, >2 or <.5). Of these genes, 614 were mapped successfully, 143 were down-regulated, and 471 were up-regulated in the aneurysms as compared with controls. Using the same criteria for significance, 3 microRNAs were identified as down-regulated and 5 were identified as up-regulated. Pathway analysis associated these genes with inflammatory response, cellular migration, and coagulation, among other functions and pathologies.
CONCLUSIONS: RNA-sequencing analysis of rabbit aneurysms revealed differential regulation of some key pathways, including inflammation and antigen presentation. ANKRD1 and TACR1 were identified as genes of interest in the regulation of matrix metalloproteinases.
ABBREVIATIONS:
- IPA
- Ingenuity Pathway Analysis
- miRNA
- microRNA
Cerebral aneurysm biology is poorly understood in general, with the mechanisms for formation, growth, healing, and rupture all in need of further elucidation. Understanding the underlying biology of aneurysms involves detailing the expression patterns of large clusters of genes and the microRNAs (miRNAs) that regulate them, alongside other biologic considerations such as hemodynamics, which involves computational modeling of the fluid dynamics of the aneurysm and surrounding vessels and has been examined in a number of recent studies.1⇓⇓⇓–5 miRNAs are small noncoding RNAs (∼20 bp) that bind messenger RNA and mediate its degradation or repression (Fig 1). Therefore, miRNA is an important regulatory molecule, and it is known to play a role in a variety of pathologies.6⇓⇓⇓⇓⇓–12 There are only a handful of previous studies that have profiled miRNA expression in either human intracranial aneurysms or relevant animal models13⇓–15 and a small number of studies that have focused on the roles of particular miRNAs.16,17 Already these studies are revealing potential biomarkers13 and the importance of these regulatory molecules.

Diagram of miRNA regulation of gene expression. mRNA indicates messenger RNA; ORF, open reading frame; Pol, polymerase; Pri-miRNA, primary microRNA; RISC, RNA-induced silencing complex.
Rabbit elastase-induced experimental aneurysms have been used to study aneurysm occlusion devices and underlying aneurysm biology.2,18⇓⇓⇓–22 These previous studies relied on detailed information about the biologic environment of experimental rabbit aneurysms. Existing studies have used RNA microarray data to quantify gene expression.18,23⇓–25 Compared with microarrays, next-generation RNA sequencing offers increased specificity and sensitivity, broader dynamic range, and the ability to detect new transcripts and isoforms.26 It also enables the detection of miRNA. In this study, we used RNA sequencing to establish differential expression patterns of messenger RNA and miRNA in experimental rabbit aneurysms.
Materials and Methods
Aneurysm Creation and Tissue Harvest
The Institutional Animal Care and Use Committee approved all procedures before initiation of the study. An elastase-induced saccular aneurysm was created in each of 6 New Zealand white rabbits by using the rabbit elastase model.27 Aneurysm and contralateral common carotid artery samples were harvested 12 weeks after aneurysm creation. These tissue samples were then immediately frozen in liquid nitrogen and stored at −70°C until they were ready for messenger RNA/miRNA extraction.
Messenger RNA and miRNA Extraction
RNA was isolated from the frozen tissues by using an miRNeasy mini kit (Qiagen, Valencia, California). The quantity of RNA was measured by using spectrophotometry, and the integrity of the RNA was confirmed by electrophoretic separation using the 2100 Bioanalyzer (Agilent Technologies, Palo Alto, California). The quality of the samples was determined by RNA integrity number.28 An RNA integrity number of >6 is considered acceptable for sequencing. One aneurysm sample that did not meet the required RNA integrity number was excluded, along with its paired control from the study, so that paired analysis could be performed on the other samples. The remaining samples (n = 5 each for the controls and the aneurysms) were used for RNA-sequencing analysis.
RNA Sequencing
RNA libraries were prepared according to the manufacturer's instructions for the TruSeq RNA sample prep kit version 2 (Illumina, San Diego, California). Then, the libraries were loaded onto paired-end flow cells following Illumina's standard protocol by using the Illumina cBot and cBot paired-end cluster kit (version 3). The flow cells were sequenced on an Illumina HiSeq 2000 using a TruSeq SBS sequencing kit (version 3) and HCS (version 2.0.12) data-collection software. Base calling was performed by using Illumina's RTA (version 1.17.21.3).
NEBNext miRNA Sequencing
miRNA libraries were prepared according to the manufacturer's instructions for the NEBNext Multiplex small-RNA kit (New England Biolabs, Ipswich, Massachusetts). Then, the libraries were loaded onto paired-end flow cells following Illumina's standard protocol by using the Illumina cBot and cBot paired-end cluster kit (version 3). The flow cells were sequenced on an Illumina HiSeq 2000 using a TruSeq SBS sequencing kit (version 3) and HCS (version 2.0.12) data-collection software. Base calling was performed by using Illumina's RTA (version 1.17.21.3).
Bioinformatics Analysis
Processing of the messenger RNA data was performed by using MAP-RSeq (version 1.2.1.3).29 MAP-RSeq consists of the following steps: alignment, quality control, obtaining genomic features for each sample, and summarizing the data across samples. The pipeline provides detailed quality-control data to estimate the distance between paired-end reads, evaluates the sequencing depth for alternate splicing analysis, determines the rate of duplicate reads, and calculates the read depth across genes by using RSeQC software (version 2.3.2).30 Paired-end reads were aligned by TopHat (version 2.0.6)31 against the April 2009 oryCun2 genome build by using the bowtie132 aligner option. Gene counts were generated by using HTSeq software (version 0.5.3p9),33 and the gene-annotation files were obtained from Ensembl (ftp://ftp.ensembl.org/pub/release-75/gtf/oryctolagus_cuniculus/Oryctolagus_cuniculus.OryCun2.0.75.gtf.gz) and the University of California Santa Cruz (http://hgdownload.soe.ucsc.edu/downloads.html#rabbit). Differential expression in a sample's aneurysm tissue compared with that in the same sample's normal tissue was computed by using the edgeR algorithm (version 2.6.2)34 across all samples. EdgeR (or empirical analysis of digital gene expression in R, an open-source programming environment) is a Bioconductor software package for examining differential expression of replicated count data; it calculates the log fold change, the P value, and the false-discovery rate between the control and experimental conditions. Human orthologs were assigned by using ExoLocator.35 The pathway analysis leveraged the Ingenuity Pathway Analysis (IPA)36 software to identify pathways enriched with human ortholog targets.
Quantitative Real-Time Polymerase Chain Reaction Analysis
First-strand complementary DNAs were synthesized from 500 ng of total RNA by using Superscript III first-strand synthesis (Invitrogen, Carlsbad, California). Real-time polymerase chain reaction assays were performed for osteopontin, von Willebrand factor, TIMP1, tyrosinase, and TACR1 with an iCycler (Bio-Rad, Hercules, California).
Statistical Analysis
The t-test statistics and corresponding P values were used as a measure of the mean change in expression between the aneurysm and control groups relative to the variability. The t-test–based P values were adjusted for multiple comparisons by using the false-discovery-rate multiple-correction approach.37 Compared with controls, genes in aneurysms with a significant difference, determined by a P value of < .05, a false discovery rate of <0.01, and a fold change of >2, were considered up-regulated, whereas those with a P value of <.05, a false discovery rate of <0.01, and a fold change of <.5 were considered down-regulated.
Results
Using the criteria discussed above for differential expression, 648 of 9396 (6.9%) genes were identified as being differentially expressed. Of these genes, 614 were mapped successfully; 143 were down-regulated, and 471 were up-regulated (On-line Table). The 34 unmapped genes consisted of rabbit genes for which no ortholog could be determined. Of 211 miRNAs measured, 11 (5.2%) mature miRNAs were identified as being differentially expressed by using the same criteria as were used for messenger RNA. Targeting information was available for 8 of them that target 230 genes that have been differentially expressed. Increased expression was seen in 5 of the miRNAs, and decreased expression was seen in 3 (Table 1).
miRNA expression data
Pathways
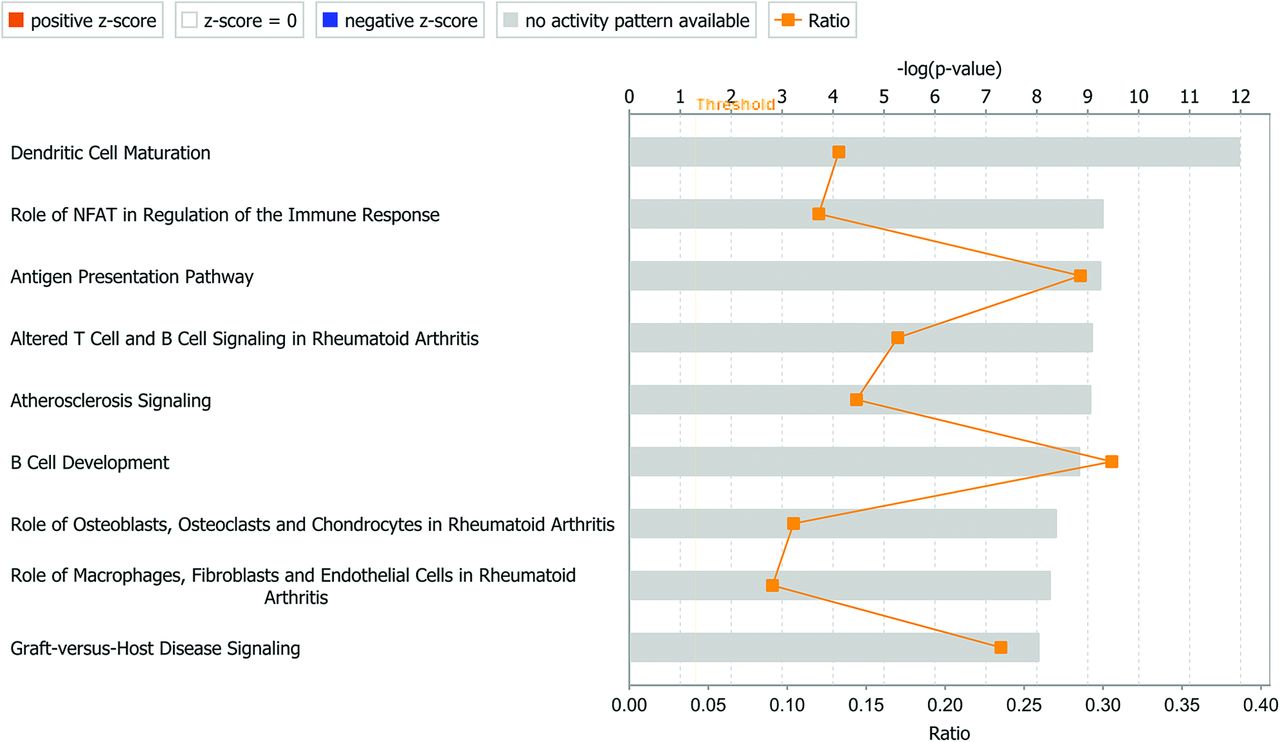
The most up-regulated pathways are shown in Table 2 and Fig 2. The first 4 pathways, all related to immune response, have the leukocyte antigen genes (HLA) in common. Another pathway of interest in aneurysms is coagulation. When comparing aneurysm tissue with control tissue, IPA identified 50 up-regulated and 12 down-regulated genes related to coagulation, which includes 9 types of collagen, with 8 being up-regulated and 1 (type 28 α1) being down-regulated (0.29-fold).
Most up-regulated canonical pathways, determined by IPA

Top 10 canonical pathways identified by IPA. NFAT indicates nuclear factor of activated T cells.
Biologic Functions of Interest
Inflammatory Response.
The inflammatory response is projected to be a pathway of major up-regulation (z score, 4.387; P = 1 × 10−31). IPA identified 65 genes with expression directions consistent with increased activity of this pathway and 21 genes with inconsistent expression directions. CC chemokine–related molecules (ligands 2 [5.3-fold]), 13 [8.7-fold], 14 [2.4-fold], 19 [7.0 fold], and 21 [8.2-fold], and receptors type 1 [32.1-fold], 2 [13.1-fold], and 5 [24.1-fold]) were up-regulated in aneurysms compared with the control arteries. The expression of interleukins was increased over the controls (ie, 7 [3.6-fold], 8 [12.4-fold], 15 [3.9-fold], and 1β [23.1-fold]), with interleukin 1 receptor type 1 also being up-regulated (2.1-fold). Caspase 1, involved in cleaving interleukin 1β into its active form, also demonstrated increased expression (11.6-fold). It is worth noting that the interleukin 1 receptor antagonist, an inhibitor of the interleukin 1 receptor, was also up-regulated in the aneurysms compared with the control (25.0-fold).
Cellular Migration.
Cellular migration is up-regulated (z score, 4.587; P = 8 × 10−43). One hundred eighteen genes with regulation directions consistent with increased migration were identified, and 61 genes with inconsistent expression directions were identified. Those involved with endothelial cell migration, which were highly expressed in aneurysms over that of the controls, included osteopontin, neuregulin 1, and fibroblast growth factor 1. Oxidized low-attenuation lipoprotein receptor 1, thrombospondin 1, and selectin E showed expression directions inconsistent with increased cellular migration in the aneurysms compared with the controls.
Validation of Microarray Data.
Verification of differential gene expression in the aneurysm and control arteries was performed for 5 selected genes. Microarray gene-expression levels were comparable with those obtained by real-time polymerase chain reactions (Fig 3).

Validation of RNA-sequencing results by real-time polymerase chain reaction assays. The real-time polymerase chain reactions are indicated by blue bars, and RNA sequencing is indicated by red bars. LCA indicates left carotid artery; OPN, osteopontin; TIMP1, TIMP metalloproteinase inhibitor 1; TYR, tyrosinase; TACR1, tachykinin receptor 1; vWF, von Willebrand factor.
Discussion
In our study we found differential expression in a large assortment of genes in tissue from experimental aneurysms compared with the contralateral common carotid arteries. The differentially expressed genes included groups related to the inflammatory response, cellular migration, and coagulation, which may provide insight into the biologic environment of unruptured human intracranial saccular aneurysms.
The pathways up-regulated in aneurysms are involved primarily in the immune response. The top 4 pathways all center around the major histocompatibility complex, which is in agreement with literature on human intracranial aneurysms.38,39
Increased expression related to inflammation was noted in human intracranial aneurysms in a number of studies.38⇓⇓⇓⇓–43 In particular, the up-regulation of genes related to the major histocompatibility complex,38,39 the complement system,41⇓–43 interleukins,42 and chemokines38,42,43 has been observed. These same inflammation markers were found in our study to be up-regulated, indicating that the rabbit saccular aneurysm model maintains fidelity to the human aneurysm with respect to inflammation.
These inflammatory molecules may be up-regulated in response to a decrease in regulatory miRNA. miR-1 and miR-204–5p were both down-regulated in the tissue. miR-1 is predicted to target CCL2 and CXCL6 and has been observed to target CXCR4,44 among others. miR-204–5p is predicted to target CCR2, CCR5, CXCR4, and IL1B, among others. miR-1 was reported to be down-regulated in human intracranial aneurysms and associated with an increased inflammatory response,14 which is in contrast to experimentally induced rat cerebral aneurysm models, which have shown increased levels of miR-1.15
Previous studies reported down-regulation of inflammatory response genes at 2 weeks in the rabbit model.24 The wider array of genes accessible via RNA sequencing in our study revealed a large number of genes with expressions consistent with increased activity of this pathway. Our results are consistent with the previous results with respect to calcium-binding glycoprotein osteonectin, which was found to be up-regulated in both studies. Although we observed a lack of histologic evidence for inflammation in the rabbit aneurysm model,45 the differential expression of inflammation-related pathways noted in this study is in accordance with that in other human studies.38⇓⇓⇓⇓–43
Krischek et al38 noted differentially regulated networks that had functions including cellular movement. In the first network they mentioned related to cellular movement, 9 of their 19 overexpressed genes were found to be overexpressed in our study as well, with none of the remaining 10 demonstrating underexpression.
ANKRD1 was the most up-regulated gene in our dataset, which may indicate that aneurysms heal via a wound-healing pathway. A dramatic increase in the expression of this gene is associated with tissue damage, and it plays an important role in the following wound-healing process.46,47 One of its modes of action is to regulate matrix metalloproteinases 13 and 10, which are involved in extracellular matrix remodeling.48 ANKRD1 has yet to receive attention in the context of aneurysm growth and healing, but its connection with matrix metalloproteinases and the wound-healing pathway, and that it was strongly up-regulated in our model, suggest that it should be a gene of interest.
TACR1 is the most down-regulated gene in our dataset, and it also relates to matrix metalloproteinase regulation. It is associated with increased expression of matrix metalloproteinase 2,49 which our group previously reported as being differentially expressed early after aneurysm formation.45 The fact that the most up-regulated and most down-regulated genes are involved in matrix metalloproteinase regulation is reflective of the fact that the role of matrix metalloproteinases in aneurysm growth, healing, and rupture is complex, being involved both for the weakening of the aneurysm wall and also the migration of endothelial cells to the neck of the aneurysm.
Our study was limited. A rabbit RNA database was used for messenger RNA expression, whereas human databases were used for miRNA because a rabbit miRNA database is not available. The RNA collected was not from a single cell type, and bias may have been introduced by the presence of cells in the aneurysm different than those in the left common carotid artery. We used IPA for pathway analysis; different software can yield different results regarding the determination of a pathway as having been up- or down-regulated. Also, “canonical” is essentially a meaningless designation, though it is the term used in the IPA software.
Conclusions
Rabbit saccular aneurysms show differential expression in a number of pathways previously reported to play roles in aneurysm biology. This expression is dominated by antigen presentation and the inflammatory response. TACR1 and ANKRD1 are genes with regulatory functions over matrix metalloproteinase activity, and their roles in aneurysm biology require further elucidation.
Acknowledgments
We thank Fariborz Rakhshan Rohakhtar, Debra A. Schultz, and Christopher Kolbert (Gene Expression Core) and Jared Evans, Daniel O'Brien, and Jamie Davila (Division of Biomedical Statistics and Informatics, Mayo Clinic).
Footnotes
Disclosures: Jennifer S. McDonald—UNRELATED: Grants/Grants Pending: GE Healthcare,* Comments: Investigator-initiated research grant for contrast-induced nephropathy. David F. Kallmes—UNRELATED: Board Membership: GE Healthcare, Comments: Cost-effectiveness board; Consultancy: ev3,* Comments: Preclinical and clinical trials; Grants/Grants Pending: MicroVention, ev3, Codman, Sequent, SurModics, and NeuroSigma, Comments: Preclinical research and clinical trials; Royalties: UVA Patent Foundation, Comments: Spine fusion. Ramanathan Kadirvel—RELATED: Grant: NIH (R01 NS076491).* *Money paid to the institution.
This work was supported by research grant NS076491 from the National Institutes of Health.
Indicates open access to non-subscribers at www.ajnr.org
References
- Received August 21, 2014.
- Accepted after revision January 30, 2015.
- © 2015 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}