
Abstract
SUMMARY: This is the second part of a retrospective and review MR imaging study aiming to define the frequency rate, timing, imaging features, and evolution of gray matter changes in Menkes disease, a rare multisystem X-linked disorder of copper metabolism characterized by early, severe, and progressive neurologic involvement. According to our analysis, neurodegenerative changes and focal basal ganglia lesions already appear in the early phases of the disease. Subdural collections are less common than generally thought; however, their presence remains important because they might challenge the differential diagnosis with child abuse and might precipitate the clinical deterioration. Anecdotal findings in our large sample seem to provide interesting clues about the protean mechanisms of brain injury in this rare disease and further highlight the broad spectrum of MR imaging findings that might be expected while imaging a child with the suspicion of or a known diagnosis of Menkes disease.
ABBREVIATION:
- MD
- Menkes disease
Menkes disease (MD; Online Mendelian Inheritance in Man No. 309400; http://omim.org/)1,2 is a rare metabolic disorder due to mutations in the ATP7A gene (Xq13.2-q13.3) encoding for the trans-Golgi copper-transporter P-type adenosine triphosphatase. Its dysfunction causes an impaired absorption and cellular metabolism of copper and results in an early, severe multisystem disease usually progressing to death in early childhood.3,4 Affected infants might exhibit a sagging facial appearance, micrognathia, arched palate, laxity of the skin (cutis laxa), supernumerary wormian bones, reduced bone density, bladder diverticula, increased vascular tortuosity, and sparse “peculiar white hair” (pili torti).5,6 Failure to thrive, hypothermia, hypotonia, impaired cognitive development, and relentless regression of developmental milestones highlight and reflect the primary involvement of the central nervous system. The scarce neuropathologic findings, both at gross and histology examination, are overtly dominated by neurodegeneration. At postmortem examination, MD typically presents with diffuse neuronal loss, gliosis, and cerebral white matter spongiosis, which result in cerebral and cerebellar atrophy.7 Consistently, in vivo brain MRIs disclose protean findings at disease onset or during the follow-up that encompass focal white and gray matter lesions and extracerebral changes but also cerebral and cerebellar atrophy, revealing that neurodegenerative changes might appear long before death. However, even though case reports and small case series can outline the mutable involvement of the brain and allow formulating possible pathogenic hypotheses of brain damage, the available literature fails to provide adequate information about the frequency, severity, and the evolution of the reported cerebral and extracerebral lesions. In addition, often the “case report” approach does not provide sufficient evidence for confirming or refuting the numerous hypothesized pathogenic mechanisms of brain involvement.
This article is the second part of an extensive retrospective and review study that investigated the neuroimaging involvement of MD. Intracranial vascular and white matter changes at onset and during the disease course have been presented in the first part. The present part will focus primarily on the neurodegenerative processes (cerebral and cerebellar atrophy), focal lesions involving the basal ganglia, and subdural collections that might be detected in these children. In addition, we will present other neuroradiologic findings that have not been reported previously, expanding the spectrum of neuroimaging findings in MD and providing additional interesting clues on the still-elusive pathogenic mechanisms leading to brain damage in this rare disease.
Materials and Methods
This section has been extensively reported in Part I.8 Briefly, MR imaging and MRA findings of 26 children with MD (mean age at first MR imaging, 7.5 ± 5.9 months; range, 0.3–32.2 months; 1 female) were retrospectively evaluated. Children were enrolled if they had a biochemically or genetically confirmed MD diagnosis and at least 1 MR imaging. Two neuroradiologists (R.M. and L.D.) with >15 years of experience in pediatric neuroradiology evaluated all MR imaging examinations (40 MRIs and 25 MRAs), aware of the diagnosis but blinded to the clinical findings; discordant findings were discussed until consensus was reached.
Parenchymal Evaluation
Qualitative parenchymal evaluation was performed in all MRIs. Cerebral atrophy was considered if there was an increase of supratentorial CSF spaces in children with normal or decreased head circumference (all our children with MD were normo- or microcephalic except patients 7 and 21). The third ventricle laterolateral diameter was measured on axial images at the level of the foramen of Monro. Cerebellar atrophy was considered when a definite enlargement of cerebellar CSF spaces was noted. Signal abnormalities of the basal ganglia, subdural collections, and any other brain signal or morphologic abnormality were also recorded.
Literature Review
Our literature review included 47 articles published between 1989 and August 2016 that reported MR neuroimaging findings in children with MD and age at MR imaging examination (On-line Appendix). As already explained in Part 1, these articles were retrieved by searching in the major databases (Embase, Scopus, PubMed, Cochrane, and also www.google.com) with the following terms: “Menkes” and “brain MR imaging.” The presence or absence of the above-mentioned MR imaging abnormalities were evaluated considering both the descriptions and available images of the selected articles. We considered as “not mentioned” any feature that could not be unequivocally defined as present or absent. We also recorded any available information about size, distribution, signal pattern, evolution, and proposed pathogenic hypothesis of MD-related brain lesions.
Statistical Analysis
The variables with normal distribution were analyzed by using the Student t test, while for ordinal variables, the Mann-Whitney U test was used. A second-order model correlation was used for fitting the third ventricle enlargement versus the age at MR imaging examination. Significance was set at P < .05.
Results
Clinical features of our sample and children in the literature with MD are summarized in the On-line Table 1, also published in Part 1. Among the 26 children with MD, 11 had died before our MR imaging evaluation (mean age, 6.3 ± 4.6 years; range, 9 months to 17.5 years). All 26 children showed early onset of severe psychomotor delay, and most of them presented with epilepsy during the early phases of MD.
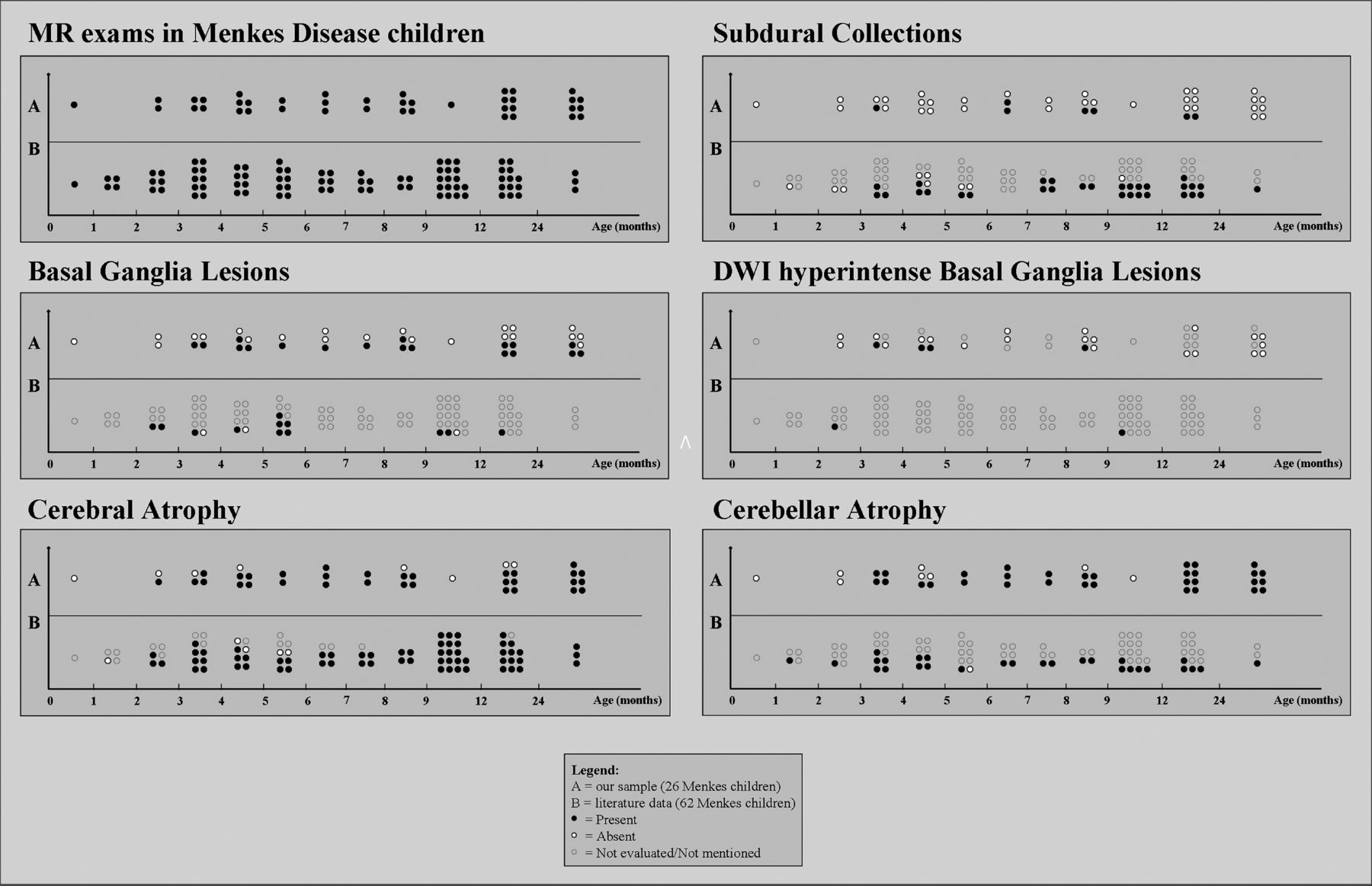
The Table and Fig 1 summarize the main neuroradiologic findings of children with MD included in the study and in our literature review.
Main neuroradiologic findings from literature review and in our sample

MR imaging findings in Menkes disease according to age at examination. A, Our sample (26 children, 40 examinations). B. Literature review (62 children, 86 examinations).
Basal Ganglia Abnormalities
Literature Review.
Basal ganglia abnormalities were detected in 11/62 children with MD (18%; mean age, 6.2 ± 4.1 months; range, 2–17 months); 1 child did not present with basal ganglia abnormalities, while the finding was not mentioned in 50/62 children. Five children with basal ganglia abnormalities had follow-up MR imaging; lesion regression or persistence was reported in 1 case each, while in 3 cases, the finding was not mentioned.
Our Sample.
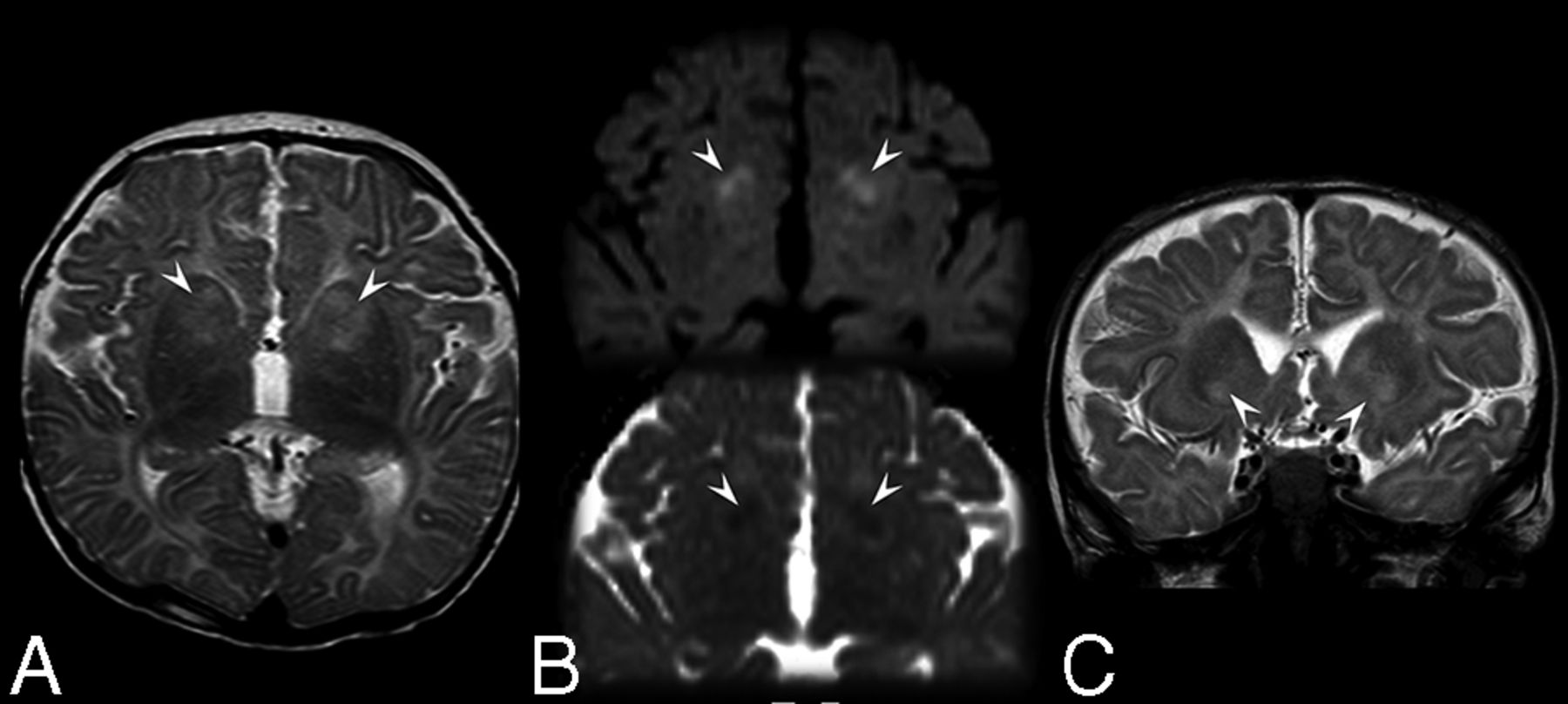
Basal ganglia abnormalities were present in 15/26 children with MD (58%; mean age, 15.7 ± 19.5 months; range, 3.6–86.7 months; Fig 2 and On-line Fig 1); in 11/15 children, the lesions were present at first MR imaging. The lesions were typically asymmetric and involved the caudate head and anterior putamen. Among children with basal ganglia abnormalities, 3 had no DWI, while in 4/12 (all younger than 1 year of age), the lesions were DWI-hyperintense with decreased ADC values. DWI ischemic-like features disappeared in 1 child, while the remaining 3/4 children had no follow-up MR imaging.

Brain MR imaging in a 5-month-old boy (patient 17) affected by Menkes disease. A, Axial T2-weighted image shows inhomogeneous hyperintensity in the anterior portion of the neostriatum more evident on the left side. B, Axial diffusion-weighted imaging (upper image) and apparent diffusion coefficient map (lower image) show small bilateral regions of restriction of the water molecule motion (arrowheads). C, Coronal T2-weighted image at the level of the anterior portion of the basal ganglia discloses the bilateral-though-asymmetric involvement of the caudate head, nucleus accumbens, and anterior putamen (arrowheads).
None of the 4 children with basal ganglia abnormalities and subsequent follow-up MR imaging showed lesion regression. Considering both the literature and our sample findings, new basal ganglia lesions were detected in the timeframe between 2 and 16 months.
Cerebral Atrophy
Literature Review.
Cerebral atrophy was detected at the first MR imaging in 41/62 children with MD (66%; mean age, 12.4 ± 21.9 months; range, 2 months to 12 years); it was absent in 5/62 and not mentioned in the remaining 16/62. At follow-up MR imaging, cerebral atrophy was reported in 21/23 children with MD (91%; mean age, 10.9 ± 6.0 months; range, 3–30 months), and it was already present, absent, or not mentioned at the previous examination in 13/21, 2/21, and 6/21 cases, respectively.
Our Sample.
Cerebral atrophy was detected at first examination in 21/26 children with MD (81%; mean age, 8.0 ± 6.2 months; range, 2.2–32.2 months) and persisted among those with available follow-up MR imaging. Regarding the 5 children with MD with no atrophy at the first examination, 2 developed it at follow-up, 2 had no follow-up, and 1 did not present with cerebral atrophy at follow-up MR imaging but the imaging was performed very early, at 3.6 months of age. Cerebral atrophy was always present in children examined after 17 months of age.
The quantitative evaluation of ventricular dilation showed a significant correlation between third ventricle diameter and age at MR imaging in male children with MD (r = 0.4, P < .001, Fig 3A). Notably, ventricle enlargement seemed to increase rapidly during the first year of life and become thereafter relatively stable. All our children with MD showed a variable increase of third ventricle diameter at follow-up (median increase, 3 mm; range, 2–9 mm; median follow-up, 10.8 months; Fig 3B).

Diameter of the third ventricle. A, Cross-sectional MR imaging findings in the 25 male children with Menkes disease show the significant correlation between ventricle dilation and age at examination (r = 0.4, P < .001). B, MR imaging findings in children with Menkes disease with neuroradiologic follow-up show significant ventricular enlargement despite a relatively short follow-up (median follow-up, 10.8 months; P = .01, paired t test).
Cerebellar Atrophy
Literature Review.
Cerebellar atrophy at first MR imaging was present in 19/62 children with MD (31%; mean age, 6.4 ± 4.5 months; range, 1.2–18 years), absent in 1/62 (5 months of age), and not mentioned in 42/62. At MR imaging follow-up, cerebellar atrophy was found in 9/23 children with MD (39%; mean age, 11.9 ± 7.5 months; range, 3–30 months), while it was not mentioned in the remaining cases; in 3/9 children with MD, the atrophy was not reported at the first examination.
Our Sample.
Cerebellar atrophy was detected at first MR imaging in 18/26 children with MD (69%; mean age, 8.7 ± 6.6 months; range, 3.1–32.2 months). In 4 children with MD, cerebellar atrophy appeared during follow-up MR imaging. All children with MD investigated after 1 year of age had cerebellar atrophy.
Subdural Collections
Literature Review.
Hematomas or hygromas at first MR imaging were present in 15/62 (24%; mean age, 7.1 ± 4.1 months; range, 3–17 months), absent in 7/62, and not mentioned in the remaining 40/62 children with MD. At follow-up MR imaging, subdural collections were present in 15/23 (65%; mean age, 12 ± 5.9 months; range, 4.5–30 months; and absent or not mentioned in the previous MR imaging in 3/15 and 7/15, respectively), absent in 2/23, and not mentioned in 6/23.
Our Sample.
Subdural collections were detected in 5/26 children with MD (18%; mean age, 9.1 ± 4 months; range, 3.6–15.6 months; On-line Fig 2). At follow-up MR imaging, subdural collections appeared in 2 further children with MD at 3.6 and 15.6 months of age and disappeared in 1 child.
Other Neuroradiologic Findings
Besides the above-mentioned findings, the thorough evaluation of the MRIs of our sample revealed several miscellaneous parenchymal abnormalities that could not be set among the known classic neuroradiologic findings of MD.
T2-Pulvinar Sign.
T2/FLAIR-hyperintensity of the posterior thalamus (Fig 4) was detected bilaterally in 4/26 children with MD, especially in advanced phases of the disease (ie, at 13.9, 14.4, 16.9, and 18.7 months of age); in 2 further children, bilateral pulvinar signal abnormalities were rather mild (8.9 and 6.6 months).

Axial FLAIR images at the level of the thalami. A, A 14-month-old boy (patient 14) shows bilateral hyperintensity of the pulvinar (arrowheads); bilateral subdural collections, brain atrophy, and abnormal myelination are also evident. B, An 87-month-old girl (patient 13) shows bilateral hyperintensity of the pulvinar. Note the severe symmetric volume loss of the thalami.
Focal Cortical-Subcortical Lesions.
The MR imaging of a 2-month-old child with MD (patient 3) presenting with repetitive seizures showed a large left DWI hyperintense cortical-subcortical parieto-occipital lesion; ADC values were decreased with the disappearance of the cortical ribbon on T1 and T2 images (Fig 5). The lesion was not recognizable at 1-month follow-up MR imaging. The MR imaging of a 4-month-old boy (patient 25) presenting with status epilepticus showed the disappearance of the cortical ribbon on T1 and T2 images in a large left parieto-occipital region that did not correspond to any vascular territory; the study protocol did not include DWI. The follow-up MR imaging performed 11 months later showed evolution into encephalomalacia of the lesion; concomitant bilateral temporal encephalomalacia was also noted.

Brain MR imaging of a 2-month-old boy (patient 3) disclosing a left cortical-subcortical occipital-temporal-parietal lesion. A, Axial diffusion-weighted images show that the lesion is markedly hyperintense (white arrowheads), mimicking an acute cerebral ischemia. ADC values were decreased (not shown). B, Coronal and sagittal T2-weighted images show the disappearance of the cortical ribbon (black arrowheads). The lesion was fully recovered at follow-up MR imaging 1 month later (not shown).
A 32-month-old boy (patient 20) showed left temporal encephalomalacia.
A 3-month-old boy (patient 21) presented with bilateral occipital cortical-subcortical T2*-hypointense lesions (posthemorrhagic encephalomalacia with hemosiderin deposits, On-line Fig 3A); the tentorium was also T2*-hypointense.
Postictal MR Imaging Changes.
The MR imaging of a 5-month-old boy (patient 17) presenting with status epilepticus revealed signal abnormalities and swelling of the left hippocampal formation (On-line Fig 3B).
A 4-month-old boy (patient 26) presenting with a status epilepticus had corpus callosum DWI hyperintensity, most likely consistent with a reversible splenium lesion (the boy had no further follow-up MR imaging to confirm this hypothesis).
Incidental MR Imaging Changes Not Directly Related to Menkes Disease
A 37-month-old boy (patient 9) underwent MR imaging a few days after cardiopulmonary resuscitation; the examination showed diffuse and severe brain ischemia with Wallerian degeneration of the corticospinal tract and trans-synaptic degeneration of the substantia nigra and the subthalamic nucleus; concomitant cortical laminar necrosis was also noted on T1 images.
A child (patient 14) presented with an iatrogenic cortical blood clot close to an intrathecal drainage set for subdural collections.
Finally, a 9-month-old boy (patient 4) had focal white matter lesions without mass effect, which were more evident in the subcortical temporal-polar regions (possible intermediate stage of transient tumefactive lesions); unfortunately, no previous or follow-up examinations were available to confirm this pathogenic hypothesis.
Further Statistical Analyses
No significant association was found between neurodegenerative changes, focal gray matter lesions, or subdural collections and clinical, vascular, or white matter findings. In particular, there was no association between basal ganglia abnormalities and tumefactive lesions and prolonged jaundice at birth or the presence or severity of intracranial artery tortuosity (P > .1 for all associations).
Discussion
The present retrospective cross-sectional multicenter study analyzed the neuroradiologic MR imaging abnormalities in a large sample of Italian children affected by Menkes disease and reviewed the pertinent literature up to August 2016. Because several children had >1 MR imaging evaluation, the study also provided interesting information on the evolution of disease-related MR imaging abnormalities. In this second section, we address the issues of neurodegeneration and basal ganglia focal lesions, comparing neuroimaging findings of our sample (26 children with MD) with those reported in the literature (62 children with MD). In addition, we discussed other previously unreported peculiar findings that widened the spectrum of MR imaging abnormalities in MD, providing interesting clues on the possible pathogenesis of brain injury.
Basal Ganglia Abnormalities
Basal ganglia involvement was common among children with MD, though the frequency rate was significantly higher in our sample than in the literature (58% versus 18%). Indeed, according to our experience, these T2/FLAIR hyperintense lesions may be easily overlooked, especially when the myelination of the surrounding white matter is impaired. Basal ganglia involvement was typically bilateral but asymmetric; the head of the caudate nucleus and anterior putamen seemed to be especially vulnerable, followed, in our sample, by the globus pallidus. According to both our cohort and literature data, the involvement of the basal ganglia was scarcely or not reversible. When the sequence was available, basal ganglia abnormalities were frequently (transiently) hyperintense in DWI; this finding implies cytotoxic edema as the main lesion mechanism for deep gray matter involvement in MD.
The detection at pathology of cystic-necrotic ischemic-like changes in the past led to hypothesizing an ischemic pathogenesis for basal ganglia lesions.7 Increased tortuosity of the main cerebral arteries could effectively result in distortion and occlusion of small perforating arteries, even in the absence of overt artery stenosis. Nonetheless, the concomitant involvement of the caudate head and anterior putamen, which are supplied by different groups of perforating arteries (originating from the anterior and middle cerebral arteries, respectively), and the lack of involvement of the interposed anterior limb of the internal capsule, which shares the same vascular supply of the head of the caudate, point to a metabolic rather than an ischemic mechanism. The resulting cytotoxic edema due to intracellular copper-related energetic failure might eventually evolve into the cystic-necrotic changes observed at pathology.
Regarding lesion timing, even though previous reports suggested a late involvement of the basal ganglia in MD,9 we found deep gray matter signal abnormalities already in the first months of life. Actually, new focal lesions were never observed after 16 months of age, pointing to early damage of these highly metabolic regions. Different from the hypotheses in previous case reports or small series,3 no significant association was found between basal ganglia abnormalities and seizures or tumefactive white matter lesions.
Cerebral Atrophy
In MD, cerebral atrophy appeared rather early during the disease course because it was recognizable in most children at first MR imaging and progressed rapidly with time, reaching a sort of “plateau” in later phases of the disease. Cerebral atrophy likely results from the combined effects of metabolic impairment, concomitant infection distress (common in children with MD), prolonged status epilepticus, increased susceptibility to oxidative stress (due to superoxide-dismutase enzyme deficiency), and energetic deficit (due to respiratory chain impairment) acting on a developing brain and resulting in severe cell death and neuronal loss.3,10⇓⇓–13 Cerebral atrophy became a constant feature at 1.5 years of age, reflecting well the strikingly dramatic neurocognitive impairment of these children. In addition, cerebral atrophy was poorly or not reversible despite the introduction, soon after disease clinical onset, of copper histidine treatment in almost all patients of our sample. Taken together, MR imaging and clinical findings confirm that neurodegenerative phenomena are of primary importance because they occur early and progress rapidly in MD. Any attempt to treat this disease should therefore take into consideration that a late treatment beginning would likely result in a poor neurocognitive outcome.
Cerebellar Atrophy
Severe depletion of cerebellar neurons (about 50% of Purkinje cells) with morphologic degenerative changes in the remaining cells14 and degeneration of all 3 cerebellar cortical layers and the deep nuclei with white matter gliosis7,10 are likely the pathologic underpinnings of MR imaging findings recorded in the present study. Indeed, the strikingly high rate of cerebellar atrophy (>80% in our sample) is consistent with the typical MD pathologic features and with the well-known clinical picture (hypotonia and cerebellar deficits are very common in children with MD). According to our experience, cerebellar atrophy was progressive, irreversible, and diffuse, with symmetric involvement of both the vermis and hemispheres. Cerebellar atrophy in MD may be easily overlooked due to the presence of other overwhelming supratentorial findings, thus explaining the difference in the detection rate between our sample and literature data. Notably, the present study showed that cerebellar atrophy was an early feature in both the literature and in our sample (1.2 versus 3 months), commonly associated with cerebral atrophy, thus highlighting the global neurodegenerative involvement of the central nervous system. Cerebellar atrophy most likely shares with cerebral atrophy its multifactorial pathogenesis and its poor response to medical treatment when started after clinical onset.
Subdural Collections
Among children with MD, subdural collections are relatively common because they might be observed in about one-fourth of cases. As repeatedly addressed in the literature, their detection at first MR imaging might raise considerable problems in the differential diagnosis with head trauma and, above all, nonaccidental head trauma.13,15⇓⇓–18 A child abuse misdiagnosis might be favored by the increased susceptibility to spontaneous fractures due to osteoporosis in MD and by the presence of supernumerary wormian bones that might mimic cranial vault fractures (On-line Fig 4). The presence of suggestive clinical (eg, “kinky hair”) or imaging features (eg, increased intracranial artery tortuosity) and specific laboratory findings (low serum copper and ceruloplasmin levels) should aid in addressing the correct diagnosis. The pathogenic mechanism leading to subdural collections is thought to depend on increased vessel wall fragility in MD,19,20 with rupture and hemorrhagic extravasation eased by the presence of brain atrophy.21 In our sample, all children with subdural collections had concomitant brain atrophy as reported in the literature, even though there was no significant association with both qualitative and quantitative evaluation of brain atrophy severity. In addition, subdural collections did not appear to be more frequent in older children with MD, when cerebral atrophy was more pronounced. Most likely, other concomitant factors such as trivial head trauma, infections, or the rapidity of atrophy progression, might have a role. From an imaging point of view, in our sample, subdural collections were extremely protean, included both hematomas and hygromas, were most frequently localized in the frontal regions, but could be hemispheric, thin, or exerting a severe mass effect on the brain structures, single or multiple, symmetric or asymmetric. Only 1 child required decompressive surgery, and the outcome was very poor.
Other Neuroradiologic Findings
Within this Part are included a few MR imaging findings that have not been clearly mentioned in previous literature but that might help in defining the whole phenotypic variability of brain damage in MD. The bilateral T2/FLAIR hyperintensity of the posterior thalamus (“pulvinar sign”) was a relatively frequent finding of MD (15%), especially in older children. Actually, subtle pulvinar involvement was also recognizable in previous studies3 and should not be unexpected because the pulvinar is a key target of prolonged seizure activity. Pulvinar cytotoxic edema is commonly detected in children with status epilepticus.22 Repetitive episodes of refractory seizures and status epilepticus characterize the clinical history of children with MD and might therefore lead to irreversible degeneration of this region. Besides, both direct and secondary trans-synaptic neurodegenerative changes might have a role, especially in the late phases of disease.23
DWI hyperintensity of the corpus callosum and swelling of the hippocampal formation found in 2 children with MD were also likely related to prolonged seizure activity. Similarly, 2 further children with MD investigated for status epilepticus had ischemic-like lesions on conventional MR imaging sequences, with large and severe cortical-subcortical involvement; one of them also had DWI that showed confirmatory cytotoxic-like features. However, 1 child showed full regression of the lesion in all sequences (including DWI), while the other, though the condition was evolving into encephalomalacia, had a nonvascular territorial involvement. Both cases therefore appeared as ischemic stroke mimics, probably related to the recent severe and prolonged seizure activity. Further evidence of a nonvascular pathogenesis is provided by the location of cortical brain injury in our sample: Two of 3 children with MD with focal encephalomalacia had lesions close to the temporal poles (1 bilaterally) (ie, in regions that are relatively protected from ischemia).
Finally, bilateral occipital cortical hemorrhagic lesions and anoxic-ischemic lesions secondary to a cardiac arrest were each identified in 1 child with MD. These findings cannot be included in the classic MR imaging abnormalities of MD. Nonetheless, they further highlight the extreme heterogeneity of brain damage among children with MD and support the hypothesis that its pathogenesis is most likely multifactorial and only partially understood.
Conclusions
Brain involvement in Menkes disease appears to be strikingly severe and heterogeneous. Besides early and poorly evolutive vascular abnormalities and delayed myelination, signs of progressive neurodegeneration are typically found. In addition, during the disease course, several cerebral and extracerebral lesions might occur, some of which are restricted within specific age windows, revealing selective age-dependent brain vulnerability during disease progression. The present detailed description of neuroradiologic findings at onset and during the disease course provides valuable clues for an early diagnosis and paves the way for the identification of reliable MR imaging biomarkers for monitoring the efficacy of new treatments. In addition, the careful analysis of lesion imaging features and evolution on a relatively large sample of patients allows dismissing pathogenic hypotheses proposed on the basis of anecdotal observations, leading to an improved definition of the possible mechanisms of brain damage in children affected by Menkes disease.
Acknowledgments
We thank the association “Angeli per la vita” and the Società Italiana di Neurologia Pediatrica, which strongly supported this spontaneous large Italian study.
The Menkes Working Group in the Italian Neuroimaging Network for Rare Diseases includes the following members: Rodica Mardari, Caterina Zanus, Gabriella Di Rosa, Consolata Soddu, Mariasavina Severino, Mario Ermani, Laura Farina, Allesandro Stecco, Gabriele Polonara, Maria Alice Donati, Lorenzo Pinelli, Carlo Dionisi-Vici, Diego Martinelli, Aba Tocchet, Giuseppe Fariello, Francesco Nicita, Daniele Frattini, Paola Martelli, and Gaetano Cantalupo.
The following are collaborators in this work: R. Mardari, Department of Neuroscience, University Hospital of Padova, Padova, Italy; C. Zanus, Institute for Maternal and Child Health, IRCCS “Burlo Garofolo,” Trieste, Italy; G. Di Rosa, Unit of Child Neurology and Psychiatry, Department of Human Pathology of the Adult and Developmental Age, University Hospital of Messina, Messina, Italy; C. Soddu, Ospedale Pediatrico Microcitemico “A. Cao,” AOB Cagliari, Italy; M. Severino, Neuroradiology Unit, Istituto Giannina Gaslini, Genova, Italy; M. Ermani, Department of Neuroscience, University Hospital of Padova, Padova, Italy; L. Farina, Department of Neuroradiology, Foundation IRCCS, Neurological Institute “C. Besta,” Milano, Italy; A. Stecco, Unit of Neuroradiology, Institute of Radiology, Department of Diagnosis, Health Services and Therapy “Maggiore della Carità,” University Hospital—Novara, Novara, Italy; G. Polonara, Clinica di Neuroradiologia, Dipartimento di Scienze Cliniche Specialistiche ed Odontostomatologiche, Universita' Politecnica delle March, Ancona, Italy; M.A. Donati, Metabolic Unit, A. Meyer Children's Hospital, Firenze, Italy; L. Pinelli, Neuroradiology, Section of Pediatric Neuroradiology, Department of Diagnostic Imaging, ASST Spedali Civili, Brescia; C. Dionisi-Vici, Division of Metabolism, Bambino Gesù Children's Hospital, IRCCS, Roma, Italy; D. Martinelli, Division of Metabolism, Bambino Gesù Children's Hospital, IRCCS, Roma, Italy; A. Tocchet, Child Neurology and Psychiatry, Department of Pediatrics and Pediatric Specialties, AOU Città della Salute e della Scienza, Torino, Italy; G. Fariello, Ospedale Fatebenefratelli, Villa San Pietro Dipartimento Immagini, Roma, Italy; F. Nicita, Department of Pediatrics and Child Neuropsychiatry, Child Neurology Division, Umberto I Hospital, Sapienza, University of Roma, Roma, Italy; D. Frattini, Child Neurology and Psychiatry Unit, Department of Pediatrics, ASMN-IRCCS, Reggio Emilia, Italy; P. Martelli, Child Neuropsychiatry Unit, “Spedali Civili,” Brescia, Italy; G. Cantalupo, Child Neuropsychiatry Unit, Department of Surgical Sciences, Dentistry, Gynecology and Pediatrics, University of Verona, Verona, Italy; F. Zennara, Institute for Maternal and Child Health-IRCCS “Burlo Garofolo,” Trieste, Italy.
Footnotes
Renzo Manara and Maria Chiara Rocco contributed equally to the study and should be considered “first coauthors”; Daniela Longo and Stefano Sartori should be both considered “senior coauthors.”
Disclosures: Renzo Manara—UNRELATED: Payment for Lectures Including Service on Speakers Bureaus: Shire, Comments: about €1000.
Indicates open access to non-subscribers at www.ajnr.org
References
- © 2017 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}