
Abstract
SUMMARY: The dentate nucleus is a cerebellar structure involved in voluntary motor function and cognition. There are relatively few entities that affect the dentate, and the clinical features of these conditions are often complex and nonspecific. Because these entities are rarely encountered, the formulation of a differential diagnosis can be difficult. Many of the conditions are reversible or treatable with early intervention. Therefore, it is important to recognize classic clinical presentations and their associated characteristic imaging findings. We provide a summary of entities that affect the dentate nucleus and a diagnostic workflow for approaching dentate nucleus imaging abnormalities.
ABBREVIATION:
- Gd3+
- free gadolinium
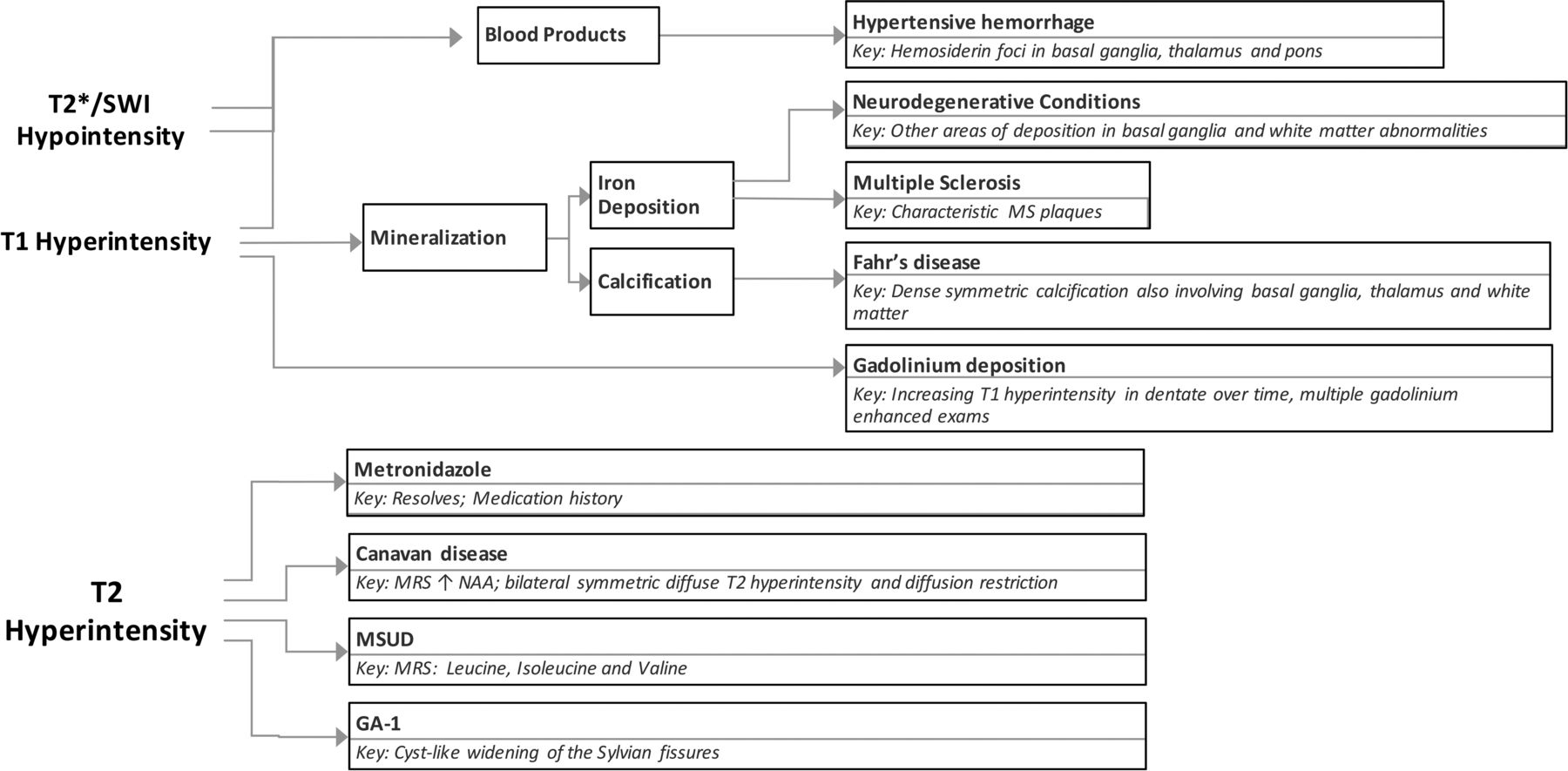
The deep cerebellar nuclei are key structures of the cerebrocerebellar circuitry that relay output from the cerebellar cortex to supratentorial cortical and subcortical targets. The dentate nucleus is the largest of the deep cerebellar nuclei, and it is involved in planning, initiating, and modifying voluntary movements as well as cognition. Because it is buried deep within cerebellar white matter, the dentate is uncommonly lesioned as a result of trauma or surgical interventions. However, a few entities specifically involve the dentate nuclei. Many of the conditions that affect the dentate are medically manageable, and prognoses can be favorable with early intervention. Despite the fact that they are rare entities, knowing the characteristic imaging findings for these conditions will allow the formulation of an appropriate differential diagnosis of dentate nucleus abnormalities and will assist in making a timely diagnosis. In this article, we review the clinical presentations and imaging features of many of the most common entities that involve the dentate. We also provide a summary of major inherited conditions and their presentations (On-line Table) and a diagnostic approach for evaluating dentate nucleus abnormalities (Fig 1).
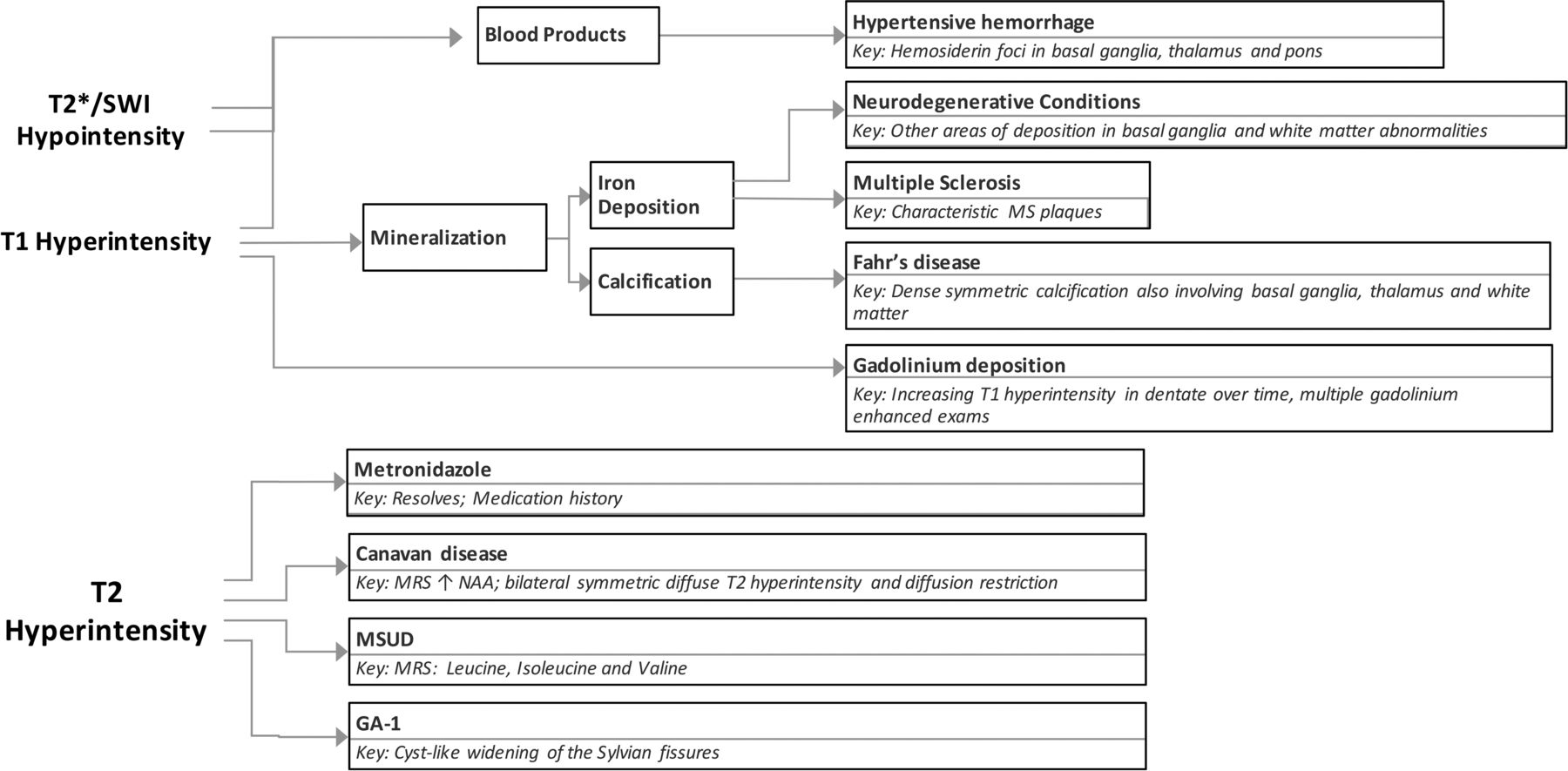
Diagnostic approach for dentate nucleus imaging abnormalities.
Normal Anatomy and Function of the Dentate
The dentate nucleus is the largest and most lateral of the 4 deep cerebellar nuclei (Fig 2). The emboliform and globose nuclei, collectively referred to as the interposed nuclei, are positioned more medially, and the fastigial nucleus lies adjacent to the midline. The dentate nucleus is located within the cerebellar white matter and is directly adjacent to the vermis and the roof of the fourth ventricle bilaterally.1,2 While the dentate is classically known to be involved in planning and executing voluntary movements, it has also been implicated in higher level cognition and sensory processing.3⇓–5 It contains anatomically separate domains that mediate these motor and nonmotor functions.6,7
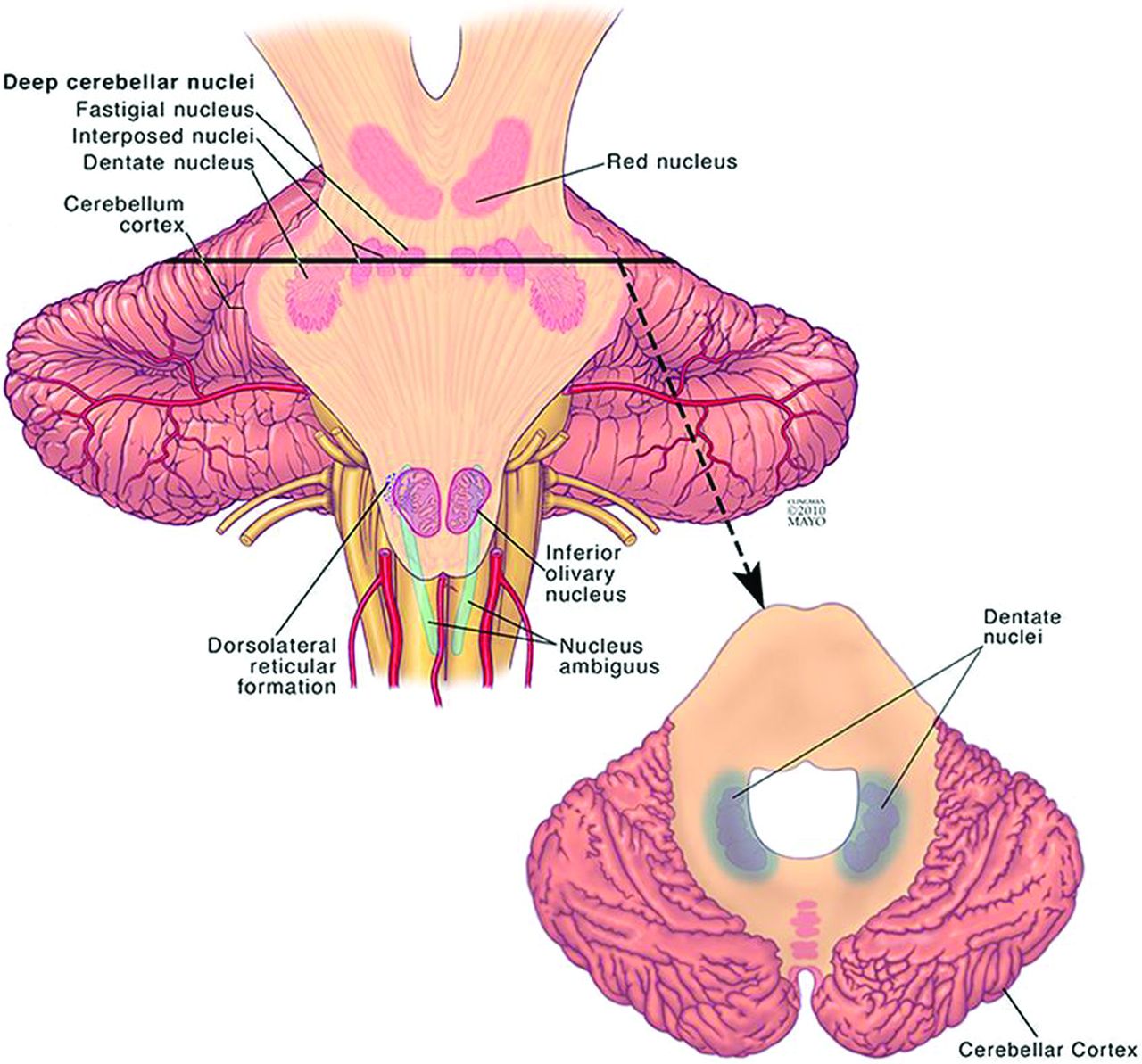
Illustration of the location of the dentate nucleus within the cerebellum. Used with permission of Mayo Foundation for Medical Education and Research, all rights reserved.
The dentate receives ascending projections from the spinocerebellar tract via the inferior cerebellar peduncle, which convey proprioceptive information about the length and tension of muscle fibers. It also receives descending projections from the premotor and supplementary motor cortices, which are involved in planning and initiating voluntary movements.2 By integrating these 2 inputs, the dentate is able to compare intended movement with muscle feedback to maintain equilibrium and balance. Its efferent projections travel through the superior cerebellar peduncle and red nucleus to the ventrolateral thalamus, where it is involved in timing and fine-tuning voluntary movements.8
It is 1 of 3 interconnected structures collectively referred to as the myoclonic triangle or the triangle of Guillain-Mollaret. This triangular circuit comprises the red nucleus, the inferior olivary nucleus, and the dentate nucleus.9,10 Parvocellular red nucleus fibers project to the ipsilateral inferior olivary nucleus by way of the central tegmental tract. Efferents of the inferior olivary nucleus travel through the inferior cerebellar peduncle to the contralateral dentate nucleus. The dentate nucleus sends inhibitory GABAergic fibers through the superior cerebellar peduncle to the contralateral red nucleus, completing the triangular circuit.11 Together, these structures control fine voluntary movement. Disruptions in the circuit that disinhibit the inferior olivary nucleus give rise to hypertrophic olivary degeneration, which can result in the clinical syndrome of palatal tremor.9
Entities Involving the Dentate Nuclei
Hypertensive Hemorrhages.
Long-standing hypertension is associated with the formation and rupture of Charcot-Bouchard microaneurysms in small penetrating blood vessels of the cerebral circulation.12 Intracerebral hemorrhage is characterized by sudden onset of a focal neurologic deficit and may be associated with headache, altered consciousness, nausea, and vomiting. Cerebellar hypertensive hemorrhage is typically caused by rupture of the perforating arteries of the superior cerebellar artery. The most common symptoms of cerebellar hemorrhage are ataxia, vertigo, severe nausea, and vomiting. Bleeding often begins in the dentate nucleus and spreads throughout the ipsilateral hemisphere to include the cerebellar peduncles and fourth ventricle.13 There is a potential for the hemorrhage to extend through the vermis to the contralateral hemisphere.
As with all hemorrhages, CT can be used to quickly identify acute bleeding. On MR imaging, T2* sequences are key to identifying areas of prior microhemorrhage and hemosiderin deposition. These T2* hypointense microhemorrhages are characteristically concentrated in the dentate, basal ganglia, thalamus, and pons (Fig 3).14
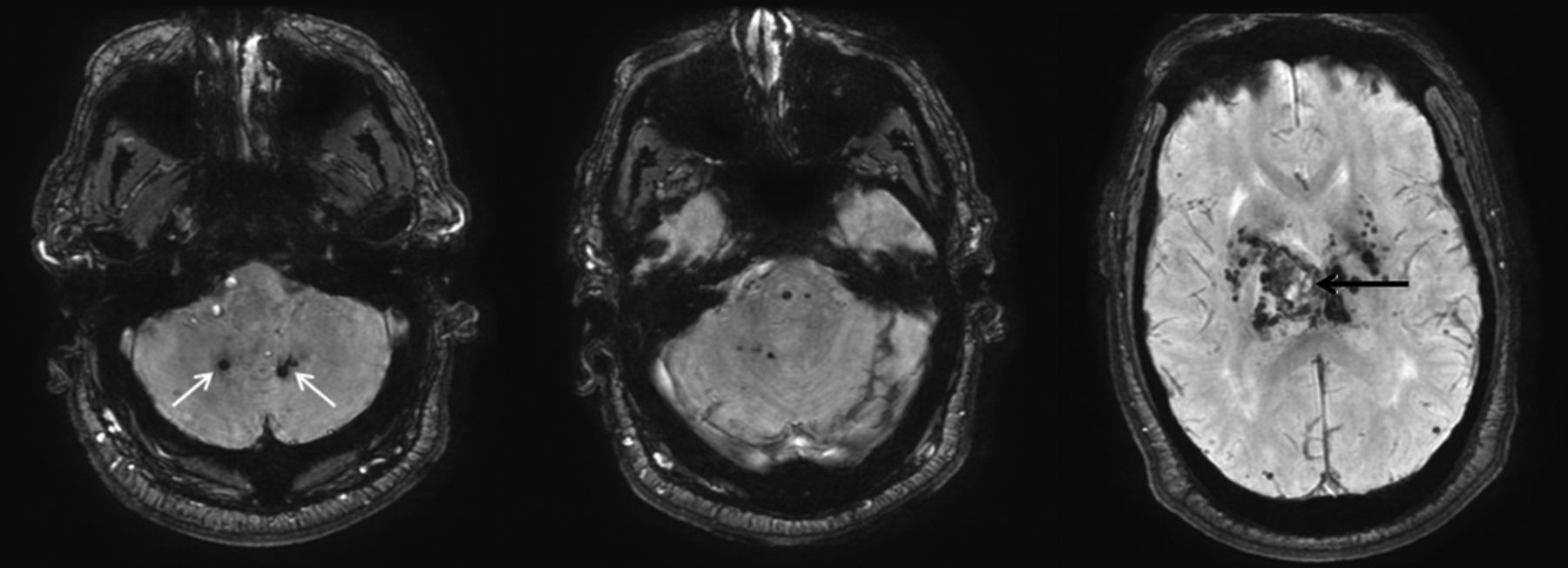
Hypertensive hemorrhage. A 55-year-old man who presented with a thalamic hemorrhage. Three selected images from 3D susceptibility-weighted imaging demonstrate microhemorrhages and hemosiderin deposition within the dentate nuclei (white arrows), brain stem, thalamus, and basal ganglia. The sentinel bleed is shown with black arrows. This distribution is characteristic of hypertensive microhemorrhages. The patient was being treated with multiple medications to control his hypertension, which remained elevated even during his hospitalization.
Transient Edema from Metronidazole.
Metronidazole is an antibiotic indicated in the treatment of a variety of bacterial and protozoan infections. While it is generally a safe medication, it crosses the blood-brain barrier and can cause serious neurologic side effects at high doses and with prolonged use. Symptoms of metronidazole toxicity include ataxia, encephalopathy, peripheral neuropathy, and seizures.15 Symptoms often resolve completely following metronidazole discontinuation.
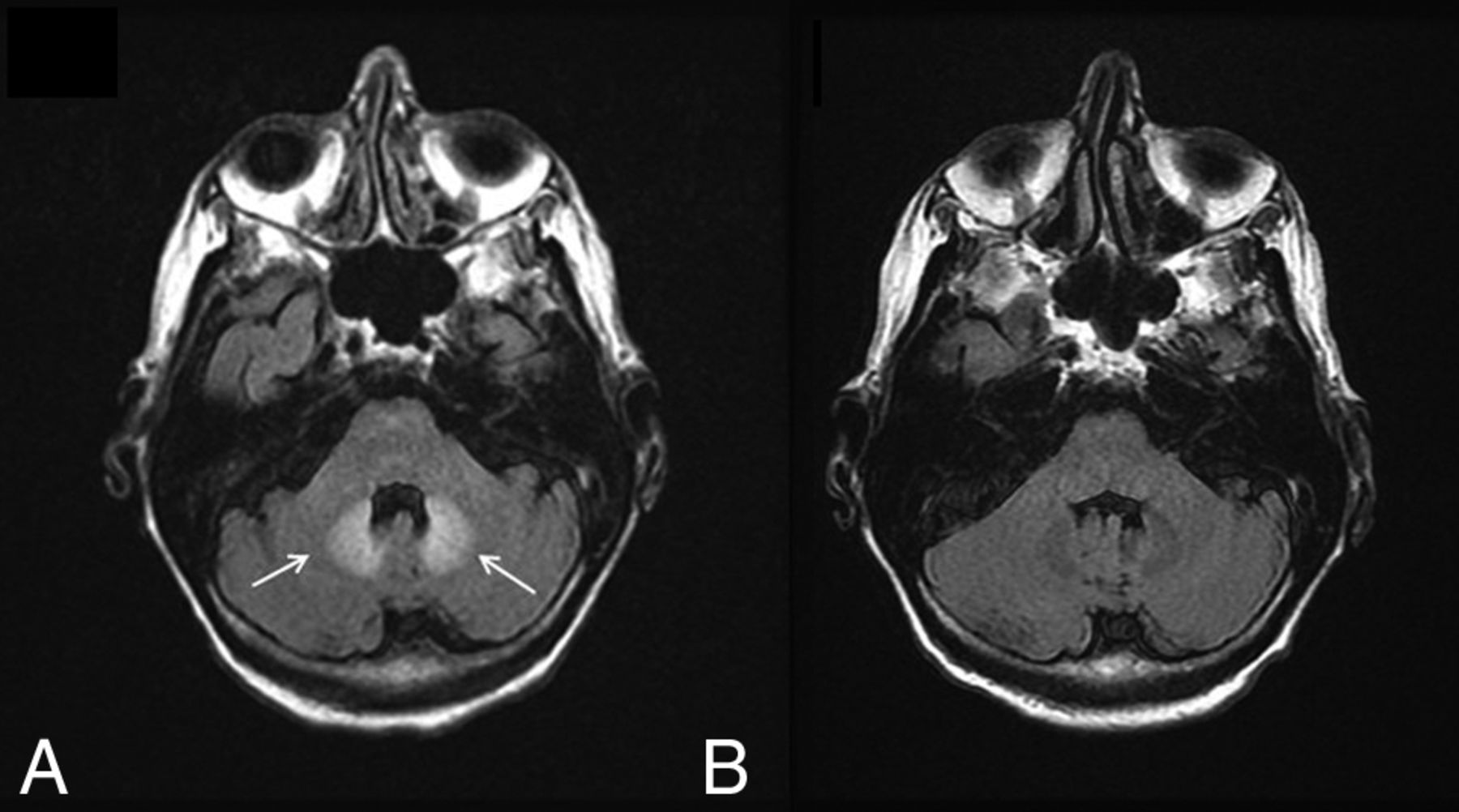
There have been reports of bilateral signal changes in the supratentorial white matter and the dentate nucleus (Fig 4) following the initiation of metronidazole therapy, both at doses within and above the recommended range.16⇓–18 These lesions are symmetric and hyperintense on T2WI and can demonstrate associated restricted diffusion. Like the symptoms associated with metronidazole toxicity, edema is reversible and imaging studies return to baseline within a few weeks of metronidazole discontinuation.18
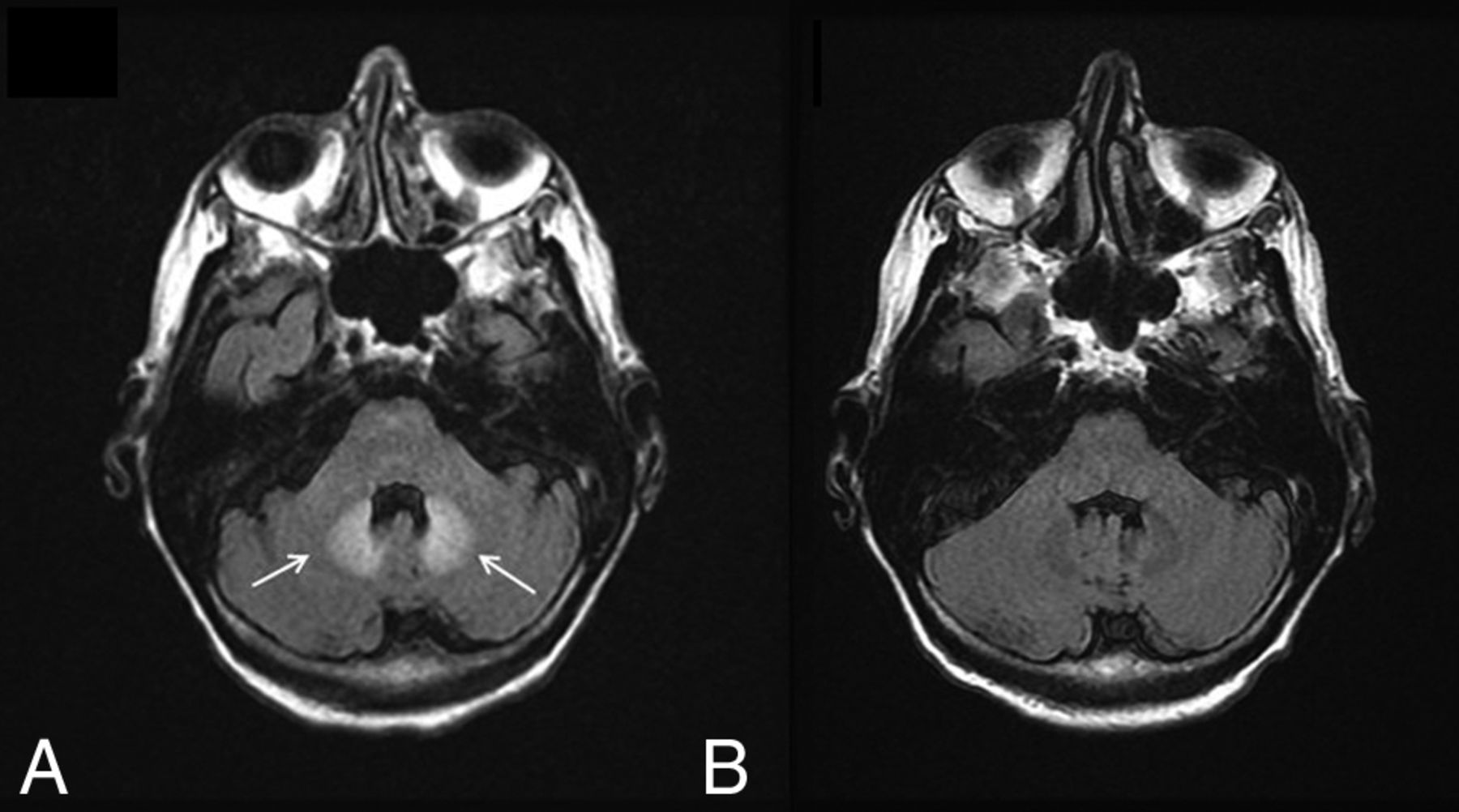
Metronidazole toxicity. A 74-year-old woman was treated for 6 weeks with metronidazole and developed dysarthria. T2 FLAIR images demonstrate T2 hyperintensity within the dentate nuclei (A, arrows), which resolved on MR imaging 1 month later (B).
Gadolinium Deposition.
Gadolinium-based contrast agents are frequently used in imaging studies. Because free gadolinium (Gd3+) is a cytotoxic element, it must be bound to a chelator that is eliminated by renal excretion when given as a contrast agent. From a safety standpoint, it is imperative that the gadolinium-chelator bond remain stable to ensure that free gadolinium is not deposited in tissues. However, Gd3+ deposition has been found in the bones of patients and animals with normal renal function following gadolinium-based contrast exposure.19
The dentate nucleus and globus pallidus have also shown high signal intensity on unenhanced T1-weighted MR imaging following multiple contrast imaging studies.20⇓–22 The number of previous contrast administrations is positively correlated with abnormal signal intensity.20 Recently, it has been confirmed that the high T1 signal in the dentate nucleus (Fig 5) and globus pallidus is due to Gd3+ deposition.23 The mechanism by which Gd3+ deposits preferentially in the dentate is not known, and to date, there have been no studies linking Gd3+ deposition to deficits in neurocognitive function or movement disorders.24

Gadolinium deposition. A 13-year-old boy was diagnosed with a pilocytic astrocytoma and treated with chemotherapy, but no radiation. A, the first available pregadolinium T1WI without any abnormality in the dentate. B, A follow-up examination performed 10 years later now demonstrates T1 hyperintensity on TIWI within the dentate nuclei (arrows). The patient had undergone 37 gadolinium-enhanced examinations at this time point.
Multiple Sclerosis.
Multiple sclerosis is an autoimmune disease of the central nervous system that affects 90 in every 100,000 individuals in the United States, with a higher prevalence in women.25 It is characterized by a cell-mediated autoimmune response against oligodendrocyte components, leading to demyelination and axonal degeneration in the brain and spinal cord. Although MS is classically known to be a disease of white matter, substantial damage and volume loss of gray matter structures can be present.26⇓–28
Prior studies have demonstrated high intrinsic T1 signal in the dentate nuclei of patients with MS. However, patients with MS tend to undergo multiple contrast-enhanced MRIs for both diagnosis and disease surveillance. Thus, the T1-weighted hyperintensity seen in the dentate nucleus in patients with MS may be a result of their large cumulative gadolinium load rather than a manifestation of their disease. Lesions demonstrating T1 hyperintensity and T2 hypointensity in deep gray matter structures, including the dentate nucleus, thalamus, striatum, and globus pallidus, can be present as well. Such lesions have been attributed to abnormal deposition of nonheme iron and evidence of MS progression.29⇓–31 It is possible that iron deposition is a by-product of neural degeneration rather than a true marker of MS progression. However, treatment with natalizumab decreases the rate of iron deposition in patients with MS, suggesting that MS-mediated inflammatory processes contribute to the phenomenon.32,33 Future studies would be needed to determine whether gadolinium or iron deposition is responsible for signal changes in the dentate of these patients.
Iron Deposition in Neurodegenerative Conditions.
There are 10 inherited neurodegenerative diseases that are characterized by iron accumulation in the basal ganglia, substantia nigra, and deep cerebellar nuclei. These rare diseases are collectively referred to as neurodegeneration with brain iron accumulation, and their combined incidence is estimated to be <1 in 1,000,000.34 Their etiologies are heterogeneous and related to defects in iron metabolism and homeostasis, cell membrane integrity, and myelin synthesis.35 The clinical symptoms of neurodegeneration with brain iron accumulation depend on which structures are involved, but many present with extrapyramidal abnormalities and cognitive dysfunction during young adulthood.34 All neurodegeneration with brain iron accumulation subtypes are progressive, and treatment consists of symptom management because there are currently no cures for the underlying disorders.
The iron deposition is best visualized on 3D susceptibility-weighted imaging as symmetric hypointensity in the affected structures. Woodhouse-Sakati syndrome is a neurodegeneration with brain iron accumulation subtype characterized by iron deposition in the globus pallidus and dentate as well as white matter abnormalities (Fig 6).
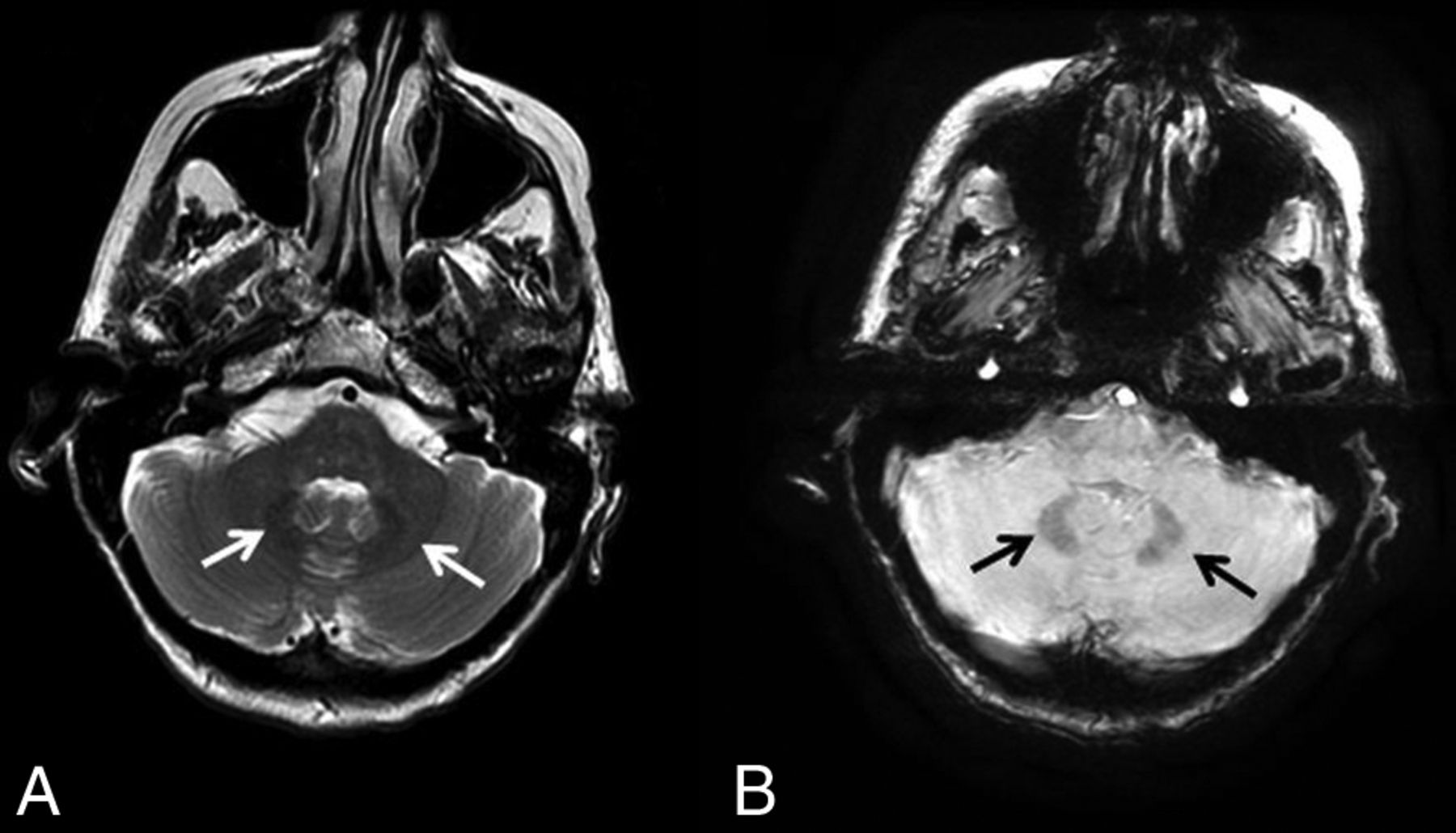
Neurodegeneration with brain iron accumulation. A 40-year-old woman with Woodhouse-Sakati syndrome found to have neurodegenerative iron accumulation. Marked hypointensity within the dentate on T2WI (A, white arrows) and 3D SWI (B, black arrows) is consistent with iron deposition.
Fahr Disease.
Fahr disease is a rare, autosomal dominant disorder that affects <1 per 1,000,000 individuals.36 It most often presents in the fourth or fifth decade of life, with progressive extrapyramidal and neuropsychiatric symptoms such as spasticity, athetosis, dysarthria, and dementia.36 While the molecular underpinnings of Fahr disease are not well-elucidated, it has been associated with a number of endocrine disorders that affect circulating calcium levels. Treatment currently aims to alleviate symptoms and manage any coexisting hormone imbalances.
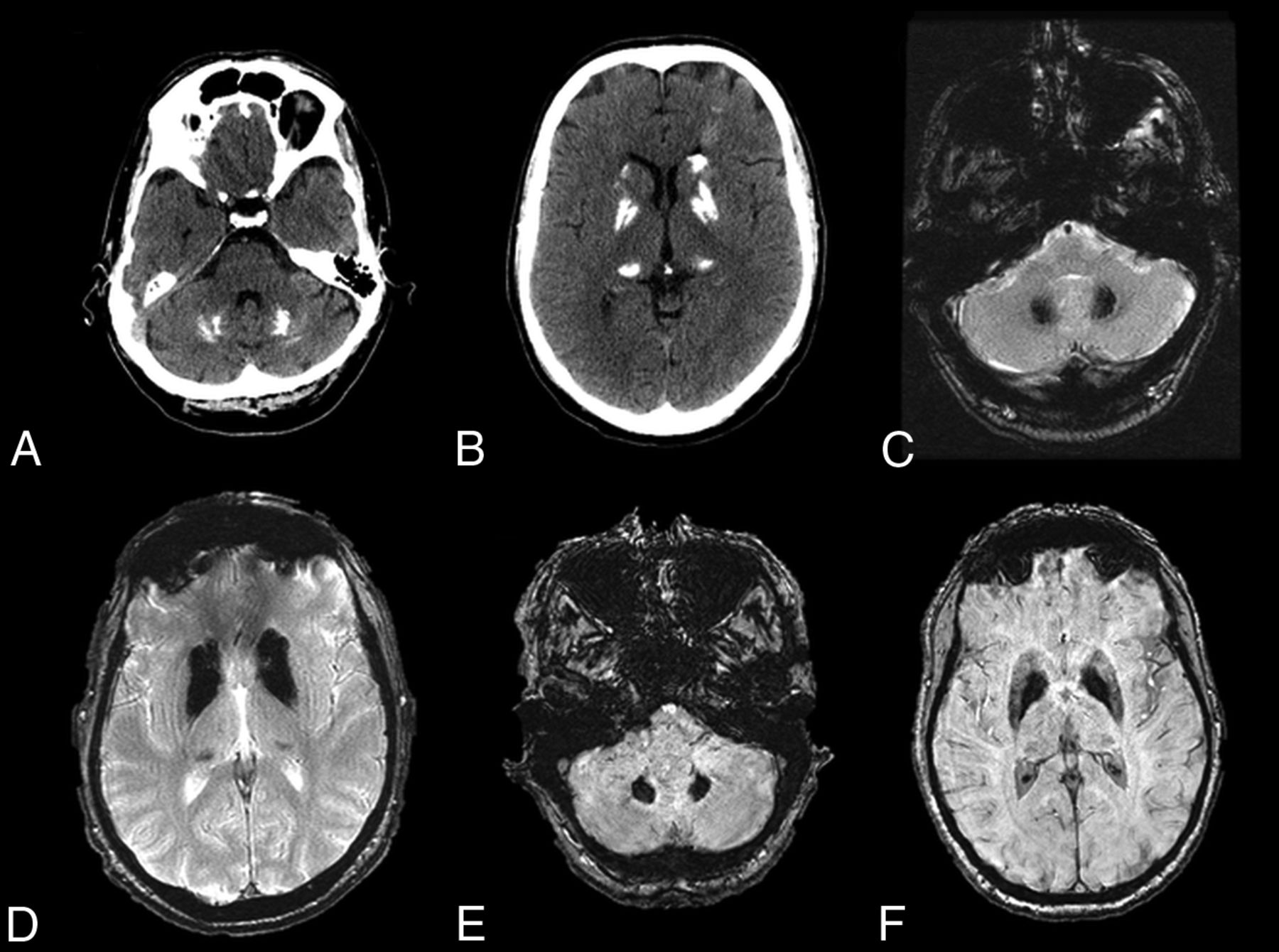
Fahr disease is characterized by idiopathic calcification of the basal ganglia and other brain regions involved in motor control, including the dentate nucleus (Fig 7) and thalamus.37⇓–39 This entity has a characteristic appearance on CT of dense, symmetric calcification most commonly in the basal ganglia, but also within the dentate nucleus, thalamus, and subcortical white matter.38 The globus pallidus is typically the first structure to be affected.36 There is no correlation between the degree of calcification and the severity of symptoms. On MR imaging, these areas of calcification can have variable T1 and T2 signal intensities that relate to the stage of disease, the degree of calcium deposition, and variance in calcium metabolism. In general, these areas will be markedly hypointense on T2*-based imaging and hypointense on T2WI and have hyperintense regions on T1WI.40 In addition, MR imaging can depict associated inflammatory changes that can later calcify and may be responsible for current symptoms.39
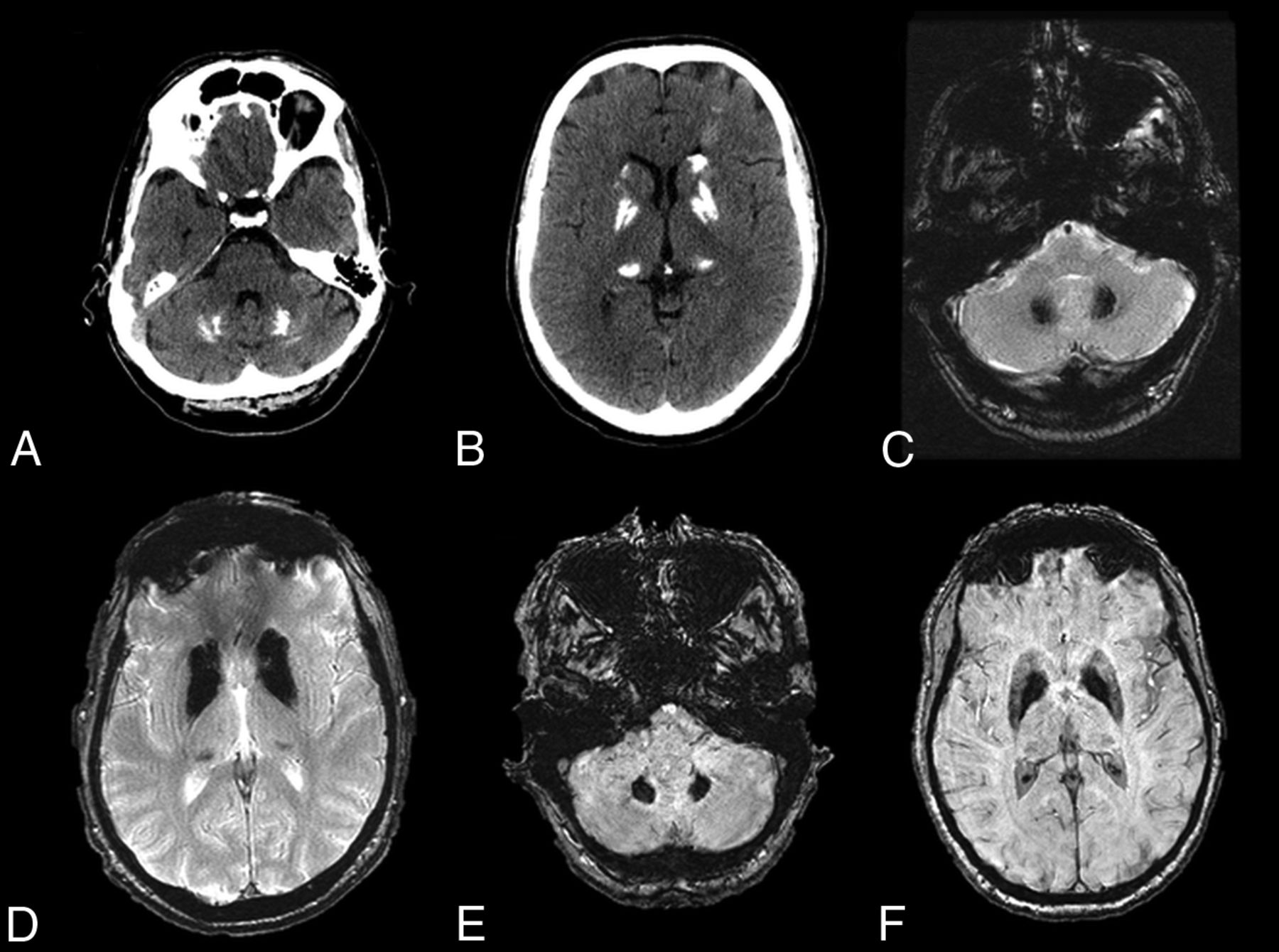
Fahr disease. A–D, CT and T2* gradient recalled-echo images from a 49-year-old patient with Fahr disease demonstrate dense calcification of the dentate nuclei and basal ganglia. E and F, Dense calcification within the dentate nuclei and basal ganglia is also well-demonstrated on susceptibility-weighted imaging, in this case from a 68-year-old affected woman.
Leigh Syndrome.
Leigh syndrome, or subacute necrotizing encephalomyelopathy, is a rare mitochondrial disease that affects 1 in 50,000 neonates.41 It is most commonly associated with a mutation in SURF1, a gene necessary for cytochrome C oxidase assembly and a functional mitochondrial respiratory chain.42 Early signs of the disease usually present within the first year of life, though it can have a late onset in adolescents and adults.43 Infants typically present with developmental delay or regression.43,44 Other signs may include seizures, dysphagia, ophthalmoparesis, nystagmus, dystonia, and ataxia. The prognosis is poor, and affected children are expected to live for only a few months after diagnosis.43,44 There is no cure for Leigh disease, and treatment is targeted at symptom management.
Leigh disease characteristically presents on imaging as symmetric T2 hyperintensity (Fig 8) and restricted diffusion of the dentate nuclei, basal ganglia, thalami, periaqueductal gray, substantia nigra, pons, and medulla.43,45,46 White matter is typically spared. The T2 hyperintensities and T1 hypointensities on MR imaging are reflective of vacuolation and spongiform changes.46 MR spectroscopy may demonstrate high levels of choline and lactate. Evidence of global atrophy can be seen as the disease progresses.46
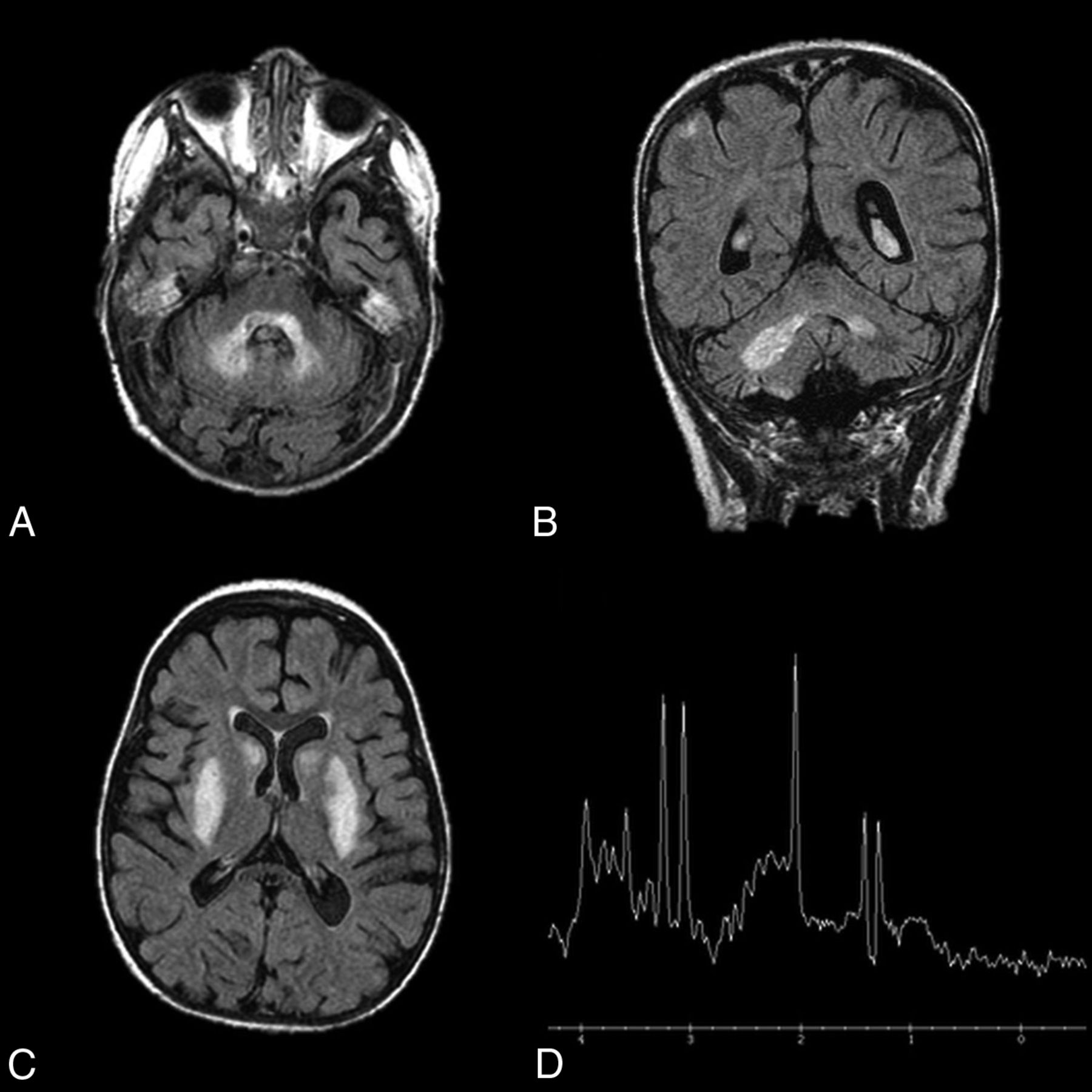
Leigh disease. An 11-year-old girl diagnosed at 2 years of age with Leigh disease. Marked T2 hyperintensity on FLAIR within the dentate nuclei (A and B) and basal ganglia (C). MR spectroscopy with voxel sampling of the basal ganglia (D) demonstrates a prominent lactate doublet (at 1.3 ppm) and elevated choline (at 3.2 ppm).
Friedreich Ataxia.
Friedreich ataxia is an autosomal recessive disease that affects approximately 2 in every 100,000 people.47 It is the most common type of inherited ataxia. Clinical presentation is variable because the disease has a variety of manifestations in the central and peripheral nervous system, heart, pancreas, and skeletal muscle. Friedreich ataxia typically presents during childhood and progresses with time, though there have been reports of onset as late as 75 years of age.47 The most common presenting symptom is progressive gait ataxia. Care is targeted at managing symptoms and addressing cardiac or endocrine manifestations of the disease.
The most common primary sites of pathologic changes in patients with Friedreich ataxia are the spinal cord and peripheral nerves.48 Thus, MR imaging consistently demonstrates atrophy of the cervical spine.49 Friedreich ataxia is also associated with selective atrophy of large neurons of the dentate nucleus with sparing of small neurons.50⇓–52 It is unclear whether the high concentration of iron in the dentate nucleus makes it particularly susceptible to disease-mediated degeneration because other iron-rich structures, such as the substantia nigra, red nucleus, and globus pallidus, are spared.50 Wallerian degeneration of the superior cerebellar peduncle is supportive of a diagnosis of Friedreich ataxia because the efferent fibers of the dentate are carried by this tract. However, involvement of these nuclei and tracts is typically not visible with conventional imaging sequences. Diffusion-weighted and diffusion tensor imaging have been used to detect these changes that are not readily apparent.53
Canavan Disease.
Canavan disease, or spongiform leukodystrophy, is an autosomal recessive disorder. The incidence of Canavan disease is estimated to be <1:100,000 in the general population and between 1:6000 and 1:14,000 in the Ashkenazi Jewish population.54,55 A mutation in the ASPA gene causes a deficiency of aspartoacyclase, resulting in a buildup of NAA, which impedes the production of new myelin and causes progressive damage to existing myelin.56,57 At postmortem examination, the white matter of brains with Canavan disease is described as edematous, gelatinous, and vacuolated, and these changes can be seen with imaging.58 Symptoms present within the first 3–6 months of life and prognosis is poor, with most affected children dying before 5 years of age.57 Treatment consists of symptom management and supportive care.
Diffuse cerebral atrophy and extensive white matter changes are characteristic of Canavan disease. The peripheral white matter is affected preferentially in the early stages of the disease.59 As the disease progresses, the globus pallidus is often atrophied, with sparing of the putamen.58,60 MR spectroscopy can be helpful in detecting elevated NAA ratios.59,61 There is variable involvement of the dentate, thalamus, brain stem, and dorsal pons. MR imaging is most helpful for evaluating the extensive white matter involvement characterized by bilaterally symmetric diffuse T2 hyperintensity and diffusion restriction.58,60
Glutaric Aciduria Type 1.
Glutaric aciduria type 1 is a rare autosomal recessive disorder that is estimated to affect 1 in every 100,000 neonates.62 In glutaric aciduria type 1, glutaric acid and 3-hydroxyglutaric acid accumulation causes neuronal hypoplasia. Glutaric aciduria type 1 usually has no signs or symptoms in the first few months of life except for microencephalic macrocephaly.63 Acute decompensation and rapid neurologic deterioration often occur following a febrile illness between 6 and 18 months of age. Symptoms include hypotonia, spasticity, dysphagia, and an increased propensity for bleeding.64 Chronic treatment involves a low lysine diet and carnitine supplementation. Many patients survive to adulthood with marked motor and coordination disability.
Even before the acute decompensation stage, patients with glutaric aciduria type 1 almost invariably have bilateral, cystlike widening of the Sylvian fissures and mesencephalic cistern, with atrophy of the frontotemporal cortices.63,65 During and after an acute decompensation event, T2-weighted hyperintensity on MR imaging and diffusion restriction on DWI are seen in the dentate nucleus, basal ganglia, periventricular white matter, and pontine medial lemniscus.65 These changes represent demyelination and edema following toxic metabolic by-product buildup. Subdural hemorrhage and hygroma can present following minor head trauma because these patients have a propensity for tearing bridging veins and arachnoid membrane fenestrations.66
Maple Syrup Urine Disease.
Maple syrup urine disease is an autosomal recessive disorder that has been estimated to affect 1 in 185,000 neonates worldwide.67 Mutations in the genes that encode for the branched-chain α-keto acid dehydrogenase complex cause a defect in the catabolism of leucine, isoleucine, and valine. If left untreated, buildup of these branched-chain amino acids and their ketoacidotic by-products can lead to rapid neurologic decline, coma, and death. With the adoption of neonate screening for maple syrup urine disease, prognosis is good in the modern world.
Untreated patients with maple syrup urine disease show no abnormalities on CT or MR imaging during their asymptomatic first few days of life.68 The 4 deep cerebellar nuclei, dorsal brain stem, cerebral peduncles, and posterior limb of the internal capsule become severely edematous about 1 week after birth at the onset of symptoms.69 Generalized edema of the brain is often superimposed on these severe, localized changes. DWI will demonstrate markedly restricted diffusion within the affected structures.69 MR spectroscopy can demonstrate abnormal peaks corresponding to increased levels of leucine, isoleucine, and valine. Edema can persist for up to 7 weeks, even if treatment is initiated promptly after the onset of symptoms. If maple syrup urine disease is treated and edema subsides, MR imaging and DWI findings rarely return to normal. Residual abnormalities include diffuse atrophy, dysmyelination, and demarcated lesions in the dentate nuclei, hypothalamus, and cerebral white matter. MR spectroscopy may show decreased, but still abnormally high, peaks corresponding to the branched-chain amino acids.69
Clues from Commonalities and Avenues for Future Research
The dentate nucleus is particularly susceptible to insults, whether demyelination, mineral deposition, or metabolic by-product buildup. The 3 other deep cerebellar nuclei and other deep gray matter structures are, for unclear reasons, often spared in these conditions. It may be that the dentate has innate biologic and metabolic characteristics that allow pathologic processes to preferentially affect it. The neurologic and pathologic effects of dentate involvement in the above-mentioned diseases have not been well-studied. Future studies examining disease-associated histopathologic changes, imaging characteristics, and natural history are needed to better characterize the pathophysiology and consequences of disease involvement of the dentate.
Conclusions
Relatively few inheritable diseases and pathologic processes affect the dentate nucleus. A summary of these diseases, their classic clinical presentations, and their radiographic features are provided in the On-line Table. The symptoms associated with these conditions can be nonspecific and complex, but the use of imaging can aid in narrowing wide differential diagnoses. Because some of these conditions are reversible or medically manageable if the diagnosis is made early enough, it is important to have a diagnostic approach for evaluating imaging features of the dentate (Fig 1).
Footnotes
Disclosures: David F. Kallmes—UNRELATED: Grants/Grants Pending: GE Healthcare, Comments: preclinical research support.* *Money paid to the institution.
Indicates open access to non-subscribers at www.ajnr.org
REFERENCES
- © 2017 by American Journal of Neuroradiology









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Structural covariation between cerebellum and cerebral cortex is atypically modulated by thalamus in autism spectrum disorder
- Effects of Gadolinium Depositions in Vivo
- Gadolinium Retention in the Brain: An MRI Relaxometry Study of Linear and Macrocyclic Gadolinium-Based Contrast Agents in Multiple Sclerosis
- Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations
- Reversible cerebellar neurotoxicity induced by metronidazole