
Abstract
SUMMARY: Biotinidase deficiency is an autosomal recessive condition caused by pathogenic variants in the BTD gene. Resultant deficiency of free biotin leads to impaired activity of the enzyme carboxylase and related neurologic, dermatologic, and ocular symptoms. Many of these are reversible on treatment, but early recognition and commencement of biotin supplementation are critical. This practice is especially important in countries where routine neonatal screening for biotinidase deficiency is not performed. In this report comprising 14 patients from multiple centers, we demonstrate the MR imaging patterns of this disorder at various age groups. Knowledge of these patterns in the appropriate clinical context will help guide early diagnosis of this treatable metabolic disorder.
Biotin is a water-soluble B-complex vitamin and is a coenzyme for 4 carboxylase enzymes that participate in gluconeogenesis, amino acid catabolism, and fatty acid synthesis.1⇓-3 The enzyme biotin holocarboxylase synthetase covalently attaches biotin to specific apocarboxylases. Proteolytic degradation of these carboxylases results in the release of biocytin and biotinyl peptides, from which free biotin is released by the enzyme biotinidase.4 Free biotin is then recycled back into the pool for use by holocarboxylases.5,6 Biotinidase also plays a role in cleaving biotin from dietary protein–bound biotin.7 Biotinidase deficiency is, therefore, characterized by an overall deficiency of free biotin and consequent defective carboxylase activity.8 Inherited as an autosomal recessive condition, biotinidase deficiency is caused by pathogenic variants in the BTD gene (Online Mendelian Inheritance in Man 609019).
Neurologic symptoms of untreated biotinidase deficiency include seizures, encephalopathy, developmental delay, vision loss, hearing loss, ataxia, and myelopathy, and these depend on the age at presentation.3,9,10 Non-neurologic features are also common, including alopecia, skin rash, and conjunctivitis. Treatment with biotin generally results in reversal of symptoms, except in cases of established hearing loss, optic atrophy, and moderate-severe developmental delay, with possible permanent deficits. Similarly, treatment with biotin before the development of clinical manifestations can prevent symptoms from developing. For this reason, routine neonatal screening for biotinidase deficiency is now performed in many countries, leading to decreased disease manifestation in these populations. In countries that lack adequate screening, however, early identification of the disease and initiation of treatment is of vital importance to prevent long-term sequelae.
Several case reports have described imaging features of biotinidase deficiency such as diffuse white matter signal changes and signal abnormalities involving the fornices, hippocampal formations, brainstem, optic pathway, and spinal cord, and these appear to be dependent on the age at presentation. In this series comprising 14 cases from multiple centers, we aimed to detail the imaging features of biotinidase deficiency presenting in various age groups.
CASE SERIES
Local institutional ethics board approval was obtained from each collaborating hospital (The Hospital for Sick Children, Toronto, Ontario, Canada; Great Ormond Street Hospital for Children, London, UK; Indira Gandhi Institute of Child Health, Bengaluru, India; Bai Jerbai Wadia Hospital for Children, Mumbai, India; and Istituto di Ricovero e Cura a Carattere Scientifico Istituto Giannina Gaslini, Genoa, Italy). Clinical, laboratory, and imaging information of 14 patients with acute presentation of biotinidase deficiency (7 males, 7 females), ranging from 2 months to 14 years of age, were retrospectively reviewed.
All MR images were acquired on a 1.5 or 3T scanner, with MR imaging sequences performed as per each hospital’s local protocol. All MR images included axial and sagittal T1-weighted, axial T2-weighted, axial or coronal FLAIR, and axial DWI sequences of the brain. MR spectroscopy was available in 1 patient, and a contrast-enhanced T1-weighted sequence was available in 1 patient. Noncontrast imaging of the cervical spine was available in 3 patients and of the whole spine in 3 patients. Spine sequences included sagittal and axial T2-weighted images, with or without T1-weighted sequences.
Clinical and MR Imaging Findings
Demographic, clinical, and imaging findings of the 14 patients are summarized in the Online Supplemental Data.
Early Infantile Group (Presentation <3 Months of Age)
Seven patients (5 male infants, 2 female infants) presented at ages ranging from 8 to 12 weeks of life (median, 10 weeks; interquartile range, 9–11 weeks), with seizures being the presenting symptom in all patients. Other features that were seen variably included developmental delay (n = 4), hypotonia (n = 3), hearing loss (n = 3), failure to thrive (n = 2), encephalopathy (n = 1), vision loss (n = 1), hyperventilation (n = 1), vomiting (n = 1), skin rash (n = 1), and alopecia (n = 1).
MR imaging in all 7 patients demonstrated diffuse cerebral and cerebellar T2 hyperintensity and swelling (Fig 1). In 2 patients (patients 3 and 10), the frontal lobe white matter was predominantly swollen compared with other regions (Fig 2). In 2 patients (patients 4 and 10), there was striking predominant involvement of the superior and inferior semilunar lobules of the cerebellum (Fig 2). Other imaging features included T2 hyperintensity at the medullary pyramids (n = 3, patients 4, 7, and 10) and perirolandic restricted diffusion (n = 3, patients 3, 4, and 7) (Fig 2).

Imaging appearance and follow-up in the early infantile age group. Axial (A and B) and coronal (C) T2-weighted images in patient 4 at 11 weeks of age at the acute stage show diffuse white matter T2 hyperintensity. D–F, The findings fully resolved on treatment with biotin.

Other imaging patterns in the early infantile age group. Coronal T2-weighted images in patient 10 at 8 weeks of age show predominant superior and inferior semilunar lobule involvement in the acute stage (A, arrow), with atrophy on follow-up MR imaging (B, arrow). C and D, Axial DWI and ADC image in patient 4 at 11 weeks of age show perirolandic restricted diffusion (arrows). Axial T2-weighted image of patient 3 in the acute stage (10 weeks of age) (E) shows predominant involvement of the frontal lobes (arrow). Axial T2-weighted image of patient 10 (F) on the follow-up image at 7 months of age shows frontal-predominant atrophy (arrow).
All patients were treated with biotin with doses ranging from 5 to 20 mg/day (median, 10 mg/day; interquartile range, 10–20 mg/day). Imaging following initiation of treatment was available in patients 4, 7, and 10 (at 2 years, 4 years, and 6 months of age, respectively), all of which showed resolution of swelling and signal abnormalities. Sequelae were only noted in patient 10 who showed frontal predominant cerebral atrophy and atrophy of the cerebellar semilunar lobules (Fig 2). On clinical follow-up, 3 patients (patients 1, 7, and 10) had sensorineural hearing loss, 2 (patients 1 and 5) had learning difficulties, and 1 (patient 10) had quadriparesis and strabismus.
Early Childhood Group (Presentation between 18 and 24 Months of Age)
Three patients (2 girls, 1 boy) presented at ages ranging from 18 to 24 months of age. All 3 patients had developmental delay and hypotonia. Two patients each had seizures (patients 2 and 6) and ataxia (patients 2 and 8). One patient (patient 6) had hearing and vision loss.
All 3 patients showed signal abnormalities in the fornices (confined only to the body in patient 2), periaqueductal gray matter, dorsal pons, and medullary pyramids. Two patients (patients 6 and 8) also had signal abnormalities in the superior and inferior colliculi; superior, middle, and inferior cerebellar peduncles; corpus callosum (inner blade of genu in patient 6, middle blade of the entire corpus callosum in patient 8); prechiasmatic optic nerves and optic chiasm; and cervical spinal cord (lateral columns in patient 6, dorsal columns in patient 8). Patient 2 also showed signal abnormalities in the medial thalami, whereas patient 6 also showed involvement of the lateral geniculate bodies, hippocampal formations, parahippocampal gyri, mamillary bodies, and hypothalamus. Imaging findings in patient 8 are depicted in Fig 3.

Imaging appearance in the early childhood age group (patient 8, 18 months of age). Axial T2-weighted images show hyperintense lesions involving the genu and splenium of corpus callosum (A, arrows), forniceal columns (A, arrowhead); dorsal midbrain including the periaqueductal gray matter (B, arrow); dorsal pons (C, arrow), medullary pyramids (D, black arrowhead), and dorsal medulla (D, white arrowhead). Axial DWI (E) and ADC (F) images show restricted diffusion involving the corpus callosum (arrows), fornices (arrowheads), and tapetum (dashed arrow). Coronal T2-weighted image (G) shows involvement of the optic chiasm (black arrowhead). Sagittal T2-weighted image of the cervical spine (H) shows faint hyperintensity involving the dorsal cervical cord (white arrowheads).
All patients were treated with biotin (10 mg in patients 2 and 8 and 15 mg in patient 6). Follow-up imaging was not available for any patient. On clinical follow-up, patients 2 and 8 recovered completely with no residual deficits, whereas patient 6 had bilateral optic atrophy and bilateral moderate sensorineural hearing loss.
Late Childhood Group (Presentation between 5 and 14 Years of Age)
Four patients (3 girls, 1 boy) presented between 5 and 14 years of age. All patients had ataxia, and 3 had myelopathy. Developmental delay was seen in 3 patients (patients 11, 12, and 13) and seizures and encephalopathy, in 2 patients (patients 11 and 12). In addition, patient 13 had regression of motor milestones. Other symptoms seen during the course of illness were vision loss (n = 3, patients 11, 12, and 14), skin rash and alopecia (n = 2, patients 11 and 14), hyperventilation (n = 2, patients 11 and 12), hearing loss (n = 1, patient 11), and vomiting (n = 1, patient 12).
Although patient 12 presented to us at 14 years of age with vision loss and myelopathy, she had a history of developmental delay, seizures, and encephalopathy at 6 months of age, at which point she was diagnosed as having a suspected metabolic disorder. No imaging was performed at this time, and she was treated with a mitochondrial cocktail of vitamins, including biotin. She developed symptoms once biotin was discontinued and was then referred for tertiary care.
The spinal cord was involved in all patients (entire cross-sectional area of the cervicothoracic cord in patient 11; lateral corticospinal tract and dorsal columns of the cervical cord in patient 12; dorsal columns of cervicothoracic cord in patient 13; and lateral and dorsal columns of the cervical cord in patient 14).
Three patients (patients 11, 13, and 14) had involvement of the fornices. In patients 11 and 13, there was symmetric forniceal involvement at onset, whereas patient 14 initially had asymmetric involvement (left > right) followed by symmetric involvement of both fornices during the next 3 months. Two patients (patients 11 and 13) had signal abnormalities in the periaqueductal gray matter, dorsal pons, and dorsolateral medulla.
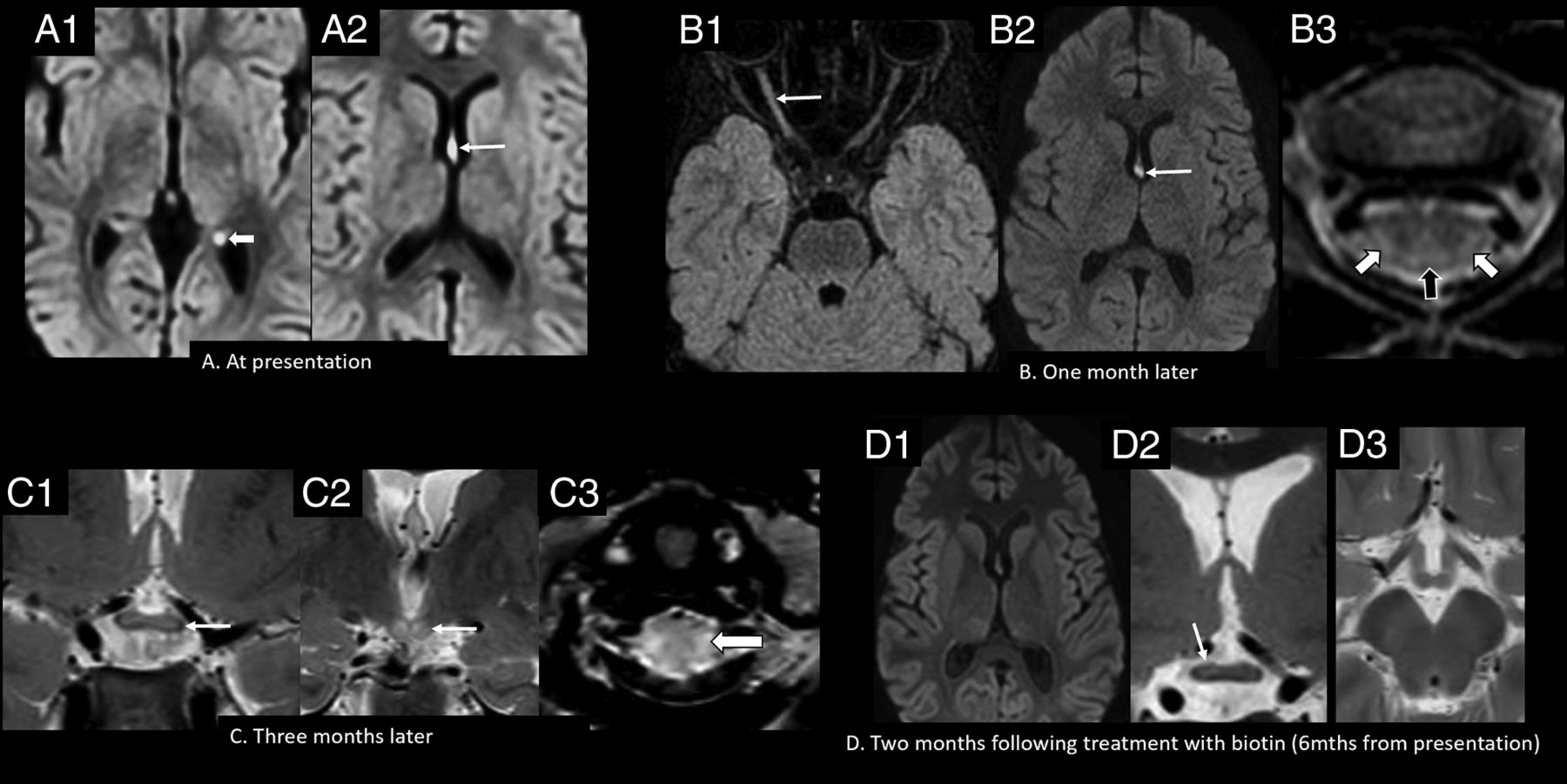
The optic pathway was involved in 2 patients. In patient 11, the prechiasmatic optic nerve and optic chiasm showed abnormal signal at onset. Patient 14, on the other hand, initially showed subtle signal change at the optic chiasm, which progressed during 3 months to symmetrically involve both optic nerves and the chiasm. Other findings included involvement of the pulvinar and hippocampal formations in patient 13 and the hypothalamus and mamillary bodies in patient 14. Imaging findings at presentation and follow-up in patient 14 are shown in Fig 4.

Imaging appearance and follow-up in the late childhood age group (patient 14, six years of age). A, Axial DWI (b-value = 1000) shows hyperintense signal involving the forniceal columns (arrow, A2) and the left forniceal crus (arrowhead, A1). Axial FLAIR (B1) and axial DWI (b-value = 1000) (B2) show hyperintense signal involving the right optic nerve (arrow, B1), with persistent signal abnormality in the fornices (arrow, B2). Axial T2-weighted image of the cervical spine (B3) shows hyperintense lesions involving the dorsal (black arrowhead) and lateral columns (white arrowheads). Coronal T2-weighted images (C1 and C2) show extension of signal abnormality to involve the optic chiasm (arrow, C1) and mamillary bodies (arrow, C2). Axial T2-weighted image of the cervical spine (C3) shows increased signal abnormality in the lateral columns (arrowhead). Axial DWI (b-value = 1000) (D1), coronal T2 (D2), and axial T2 (D3) weighted images show resolution of the forniceal and mamillary body hyperintensity, with faint residual signal change in the optic chiasm on the right (arrow, D2). Mths indicates months.
All patients were treated initially with 20 mg of biotin. The dose of biotin was gradually increased to 80 mg/day in patient 14, due to worsening of symptoms, and this dose has been continued since. Follow-up details for 1 patient (patient 13) were not available. The other 3 patients had residual symptoms at follow-up, with strabismus in patients 11 and 12 and rash, mild distal hypotonia and muscular weakness, and ataxia in patient 14.
Follow-up imaging following initiation of biotin therapy was available in patients 11 and 14. Both patients showed resolution of the intracranial signal abnormalities (Fig 4) but residual signal changes in the spinal cord.
DISCUSSION
All patients in the early infantile group showed diffuse cerebral and cerebellar white matter swelling and abnormal T2 hyperintense signal on imaging during presentation. These are congruent with the findings of other studies describing a similar pattern in patients presenting between 4 weeks and 5 months of age.11⇓⇓-14 Cases with a predilection for the frontal and frontoparietal lobes have also been described,12 similar to 2 cases in our series. We, however, did not find any reports describing predominant involvement of the superior and inferior semilunar lobules of the cerebellum, as was seen in 2 of our cases. Similarly, T2-hyperintense signal at the level of the medullary pyramids has not been previously described.
Various hypotheses that have been proposed for diffuse white matter signal abnormality in infants with biotinidase deficiency include interstitial edema,11,12,14 delayed myelination,11 dysmyelination,12 ischemia,11 and leukodystrophy.13 Given the presence of swelling and striking T2 hyperintensity, we believe that the main process driving these changes is interstitial edema rather than leukodystrophy or dysmyelination. The diffuse pattern of involvement (involving both myelinated and unmyelinated white matter) and the reversibility of changes in most patients in our series, and others12,15⇓-17 favor this hypothesis. Atrophy on follow-up imaging was seen in only one of our patients, involving the frontal lobes and cerebellar semilunar lobules, suggesting a degree of irreversible injury in this patient by the time treatment was initiated.
Another finding we present in 2 of our patients in the early infantile age group is restricted diffusion in the perirolandic regions. Soares-Fernandes et al14 described a strikingly similar case and attributed it to accelerated myelination, possibly induced by the repeat neuronal electrical activity of seizures. This may be plausible, given that all patients in our series in this age group presented with seizures. Also, early regions of myelination have increased metabolic activity/requirements and therefore might be more susceptible. Interestingly, a few case reports have described striking, restricted diffusion in the internal capsules, optic radiations, corticospinal tracts of the brainstem,18⇓-20 splenium of corpus callosum,18,20 cerebellar white matter,18,20 and the hippocampi and medial temporal regions,19⇓-21 none of which were observed in our series. Most of these patients presented at, or were imaged between 3 and 8 months of age, and whether this differential pattern therefore relates to the timing of metabolic decompensation is a matter of debate.
In our series, there was considerable overlap in imaging features in the early and late childhood age groups. The unifying finding in all 3 patients in the early age group was T2 hyperintensity in the fornices, periaqueductal gray matter, dorsal pons, and medullary pyramids, with variable involvement of the corpus callosum, cerebellar peduncles, optic pathway, diencephalic structures, and cervical spinal cord. Restricted diffusion of the fornices was seen in all 3 patients, whereas the other structures showed variable features on DWI. Forniceal, diencephalic, and brainstem involvement has previously been reported in various age groups, ranging from 3 years to adulthood,16,17,22⇓-24 whereas only 1 case describing this pattern was found in toddlers younger than 2 years of age (case 1, Mc Sweeney et al22). Similarly, spinal cord and optic chiasm involvement have been described infrequently in children 3 years of age and older,15⇓-17,21,23⇓-25 whereas 2 of our patients younger than 2 years of age showed variable involvement of these structures. Interestingly, in 1 case each, there was involvement of the tapetum and lateral geniculate bodies, respectively, both not previously described.
The imaging finding common to all patients in the late childhood age group was spinal cord involvement, with variable involvement of the other structures that were also involved in the early childhood age group. In those with spinal cord involvement, the cervical cord was involved in all patients across both groups, and this involvement is in keeping with the findings of other series.15⇓-17,22⇓⇓⇓-26 Thoracic or holocord involvement contiguous with cervical cord abnormality has also been commonly described15,17,21⇓⇓-24,26 but was seen only in 2 of our patients across both groups. In those with spinal cord involvement, only one of our patients showed involvement of the entire cross-sectional area, whereas the others showed selective involvement of certain columns, with the dorsal column being commonly involved. Other authors have shown more common involvement of the entire cross-sectional area,15,26 though selective column involvement has also been described.15,21,24,25 The reason for this differential involvement is unknown. Optic pathway involvement in our series was confined to the optic nerves and chiasm, similar to findings in other reports.17,24 Additional previously described components of the optic pathway include the optic tracts23 and optic radiations.18,19
Similar to findings in other studies,12,15⇓-17 there was clinical and radiologic improvement in all patients following initiation of treatment. The most common residual deficits after treatment in our series included sensorineural hearing loss and visual impairment, the latter attributable to optic atrophy. Irreversibility of established hearing and vision loss is well-known in biotinidase deficiency.27 Other residual deficits in our series and others15,23,24,26 included learning difficulties, ataxia, hypotonia, and quadriparesis.
Imaging-based differential diagnoses for biotinidase deficiency vary depending on the pattern of involvement. For instance, diffuse white matter T2 hyperintensity and swelling are nonspecific and may be seen in urea cycle defects, organic acidemias, and amino-acidemias presenting with metabolic decompensation.28 Recently, forniceal restricted diffusion was demonstrated to be present in 53/714 (7.4%) children younger than 2 years of age.29 The authors postulated this to be a transient abnormality possibly related to seizures, though only 4/53 patients had follow-up imaging showing resolution. It is unclear whether any of these children were screened for biotinidase deficiency. Finally, signal alterations in the optic chiasm, brainstem, and spinal cord should prompt the consideration of demyelinating disorders such as myelin oligodendrocyte glycoprotein antibody disease and neuromyelitis optica spectrum disorder in the appropriate clinical setting.30
Given the dramatic impact of neonatal screening and treatment before development of symptoms,31 screening for biotinidase deficiency is now offered in >30 countries worldwide.32 Most countries, however, do not routinely screen for this disease; early recognition and treatment are, therefore, crucial to prevent long-term sequelae.
CONCLUSIONS
Recognizable imaging features of biotinidase deficiency at different age groups include a predilection for diffuse white matter edema in young infants and a predilection for the involvement of the fornices, diencephalic structures, optic pathway, brainstem, and spinal cord in toddlers and older children.
Footnotes
S. Batzios and K. Mankad are joint senior authors.
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received September 26, 2022.
- Accepted after revision January 4, 2023.
- © 2023 by American Journal of Neuroradiology





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.