
Abstract
BACKGROUND AND PURPOSE: Loeys-Dietz syndrome (LDS) is a recently described entity that has the triad of arterial tortuosity and aneurysms, hypertelorism, and bifid uvula or cleft palate. Its neuroradiologic manifestations have not been well delineated. We sought to describe the neuroradiologic features of LDS and to assess the manifestations that would warrant follow-up imaging.
MATERIALS AND METHODS: Two neuroradiologists retrospectively reviewed CT angiography (CTA), MR imaging, and plain film studies related to the head and neck in 25 patients ranging from 1 to 55 years of age, all of whom had positive genetic testing and clinical characteristics of LDS. Arterial tortuosity was evaluated by subjective assessment of 2D and 3D volumetric CTA and MR angiography data. Craniosynostosis and spinal manifestations were assessed by using plain films and CT images. MR images mostly of the head were reviewed for associated findings such as hydrocephalus, Chiari malformation, etc. Clinical manifestations were collated from the electronic patient record.
RESULTS: All patients had extreme arterial tortuosity, which is characteristic of this syndrome. Thirteen patients had scoliosis, 12 had craniosynostosis, 8 had intracranial aneurysms, 6 had spinal instability, 3 had dissections of the carotid and vertebrobasilar arteries, 3 had hydrocephalus, 4 had dural ectasia, 2 had a Chiari malformation, and 1 had intracranial hemorrhage as a complication of vascular dissection.
CONCLUSIONS: Significant neuroradiologic manifestations are associated with LDS, predominantly arterial tortuosity. Most of the patients in this series were young and, therefore, may require serial CTA monitoring for development of intra- and extracranial dissections and aneurysms, on the basis of the fact that most of the patients with pseudoaneurysms and dissection were older at the time of imaging. Other findings of LDS such as craniosynostosis, Chiari malformation, and spinal instability may also need to be addressed.
The Loeys-Dietz syndrome (LDS) was first described in 2005 as autosomal dominant with widespread systemic involvement (OMIM, On-line Mendelian Inheritance in Man, # 609192; http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim). The disease is characterized by the triad of the following: 1) arterial tortuosity, aneurysms, or dissections, 2) hypertelorism, and 3) bifid uvula or cleft palate.1,2 It is caused by heterozygous mutations in the genes encoding transforming growth factor B (β) receptors (TRGFBR) 1 or 2 with TGFBR1 traced to chromosome 9q33–q34 and TGFBR2 to chromosome 3p24.
Affected patients have a high risk of arterial dissection or rupture at an early age and require early aortic root replacement surgery.2 Some individuals have phenotypes that overlap Marfan syndrome.2,3 In patients with LDS, neurovascular aneurysms and dissections are particularly prevalent, and these may have complications in the central nervous system (CNS) that are not otherwise associated with this syndrome.1–3
To our knowledge, the neuroradiologic manifestations of the LDS have not as yet been described in the neuroradiology literature. We sought to define the risks of aneurysms and dissections in the carotid and vertebrobasilar system as well as skeletal findings in the spine and skull and to assess the findings that warranted follow-up imaging for manifestations of this syndrome.
Materials and Methods
We retrospectively reviewed 25 patients with the diagnosis of LDS based on a combination of clinical criteria and genotyping. Patients ranged from 1 to 55 years of age (mean, 18.2 ± 15.7 years), and there were 12 female and 13 male patients. All patients had positive genetic testing for LDS and had craniofacial anomalies associated with LDS type I.
The study was approved by the institutional review board as a retrospective study. Two neuroradiologists reviewed the CT, CT angiography (CTA), MR imaging, and plain films of the head, neck, and spine in these patients for various neuroradiologic manifestations. 3D reconstructions were performed on the CTA studies. All studies were performed either on a 64-section multidetector row CT (MDCT) scanner (Sensation 64 and Definition; Siemens, Erlangen, Germany) or a 16-section MDCT scanner (Aquilon 16; Toshiba Medical Systems, Tokyo, Japan).
Parameters for the CTA acquisition were 0.75-mm section thickness, 3-mm table feed per rotation, 0.33-second gantry rotation time, a pitch of 0.55, 120 kV, 340 mAs, 512 × 512 matrix, and 25-cm FOV (varying according to patient size) on the Siemens scanner and 0.5-mm section thickness, 3-mm table feed per rotation, 0.5-second gantry rotation time, a pitch of 0.75, 120 kV, 300 mAs, 512 × 512 matrix, and 25-cm FOV (varying according to patient size) on the Toshiba scanner. Lower doses of milliampere-second were used in pediatric patients. The volumetric data were transferred to a Siemens and/or Vitrea 2 workstation (Vital Images, Minnetonka, Minn), and 2D multiplanar and 3D reconstructions were performed by using both the Siemens InSpace and Vitrea 2 commercially available software.
MR imaging of the brain was performed on Verio, Trio, Avanto, and Espree (Siemens) and Intera and Achieva (Philips Medical Systems, Best, the Netherlands) 1.5T and 3T magnets with standard protocols in the axial, coronal, and sagittal planes by using T1-weighted, T2-weighted, fluid-attenuated inversion recovery, MR angiographic (MRA), and postcontrast T1-weighted images. Spine MR imaging studies used sagittal T1-weighted, T2-weighted, and short τ inversion recovery (STIR) sequences with axial gradient-echo and T2-weighted scans. Head and neck MR imaging studies relied on axial T1-weighted and axial STIR sequences with sagittal T1-weighted images. Postcontrast scans were variably obtained. Plain films of the skull and spine were obtained in the anteroposterior and lateral projections. Flexion extension views were obtained for evaluation of spinal instability.
CTA images were reviewed for aneurysms, dissections, pseudoaneurysms, and arterial tortuosity. Associated CT images were reviewed for hydrocephalus, craniosynostosis, arachnoid cysts, and Chiari malformation, which is infrequently associated with this syndrome. MR images were reviewed for associated findings like hydrocephalus, craniosynostosis, arachnoid cysts, and Chiari malformation. Plain films and CT of the spine were reviewed to evaluate dural ectasia, spinal instability, and scoliosis. All patients with a diagnosis of LDS at our institution underwent CT of the head, neck, chest, abdomen, and pelvis. CTA of the head and neck was also performed on all the patients in our series. Reformatted images were obtained to evaluate the spine, and MR imaging was performed if neuroradiologic findings on CT mandated an MR imaging for further evaluation.
The craniofacial severity index was assessed by clinical collaborators and was used to determine the severity of symptoms of LDS. Scores ranged from 1 to 11 based on the severity of hypertelorism, bifid uvula, cleft palate, and craniosynostosis.
Craniofacial Severity Index
The craniofacial severity index is used to determine the severity of symptoms of the LDS and has been observed to correspond to aortic root measurements and cardiovascular outcome.2 The index is scored on a scale of 0–11 and is graded by the status of hypertelorism (none = 0, subtle [interpupillary distance at or around the 97th percentile] = 1, and marked = 2), appearance of the uvula (normal = 0, broad without cleft = 1, midline raphe = 2, bifid uvula = 3), and cleft palate or craniosynostosis (none = 0, 1 = 3, both = 6).
Results
All of the patients were classified as LDS type 1 by the clinical team on the basis of genetics, craniofacial findings, and clinical examinations.
Imaging Manifestations
Review of the CTA studies revealed the following imaging manifestations.
Vascular, Brain, and Skull
All 25 patients had CTA performed as part of their evaluation (Figs 1⇓–3). Eight (32%) of the 25 patients had aneurysms (7 intracranial, 1 extracranial). Six aneurysms were located in the internal carotid artery (ICA) or its branches, and 2 were along the vertebrobasilar system. Aneurysms ranged from 1 to 8 mm. Three (12%) of the 25 patients had either intra- or extracranial dissections and/or pseudoaneurysms of the internal carotid, vertebral, or subclavian arteries. Dissections were both short- and long-segment. All 25 patients had arterial tortuosity, some extreme. One patient had intracranial hemorrhage as a complication of cerebral artery dissection.
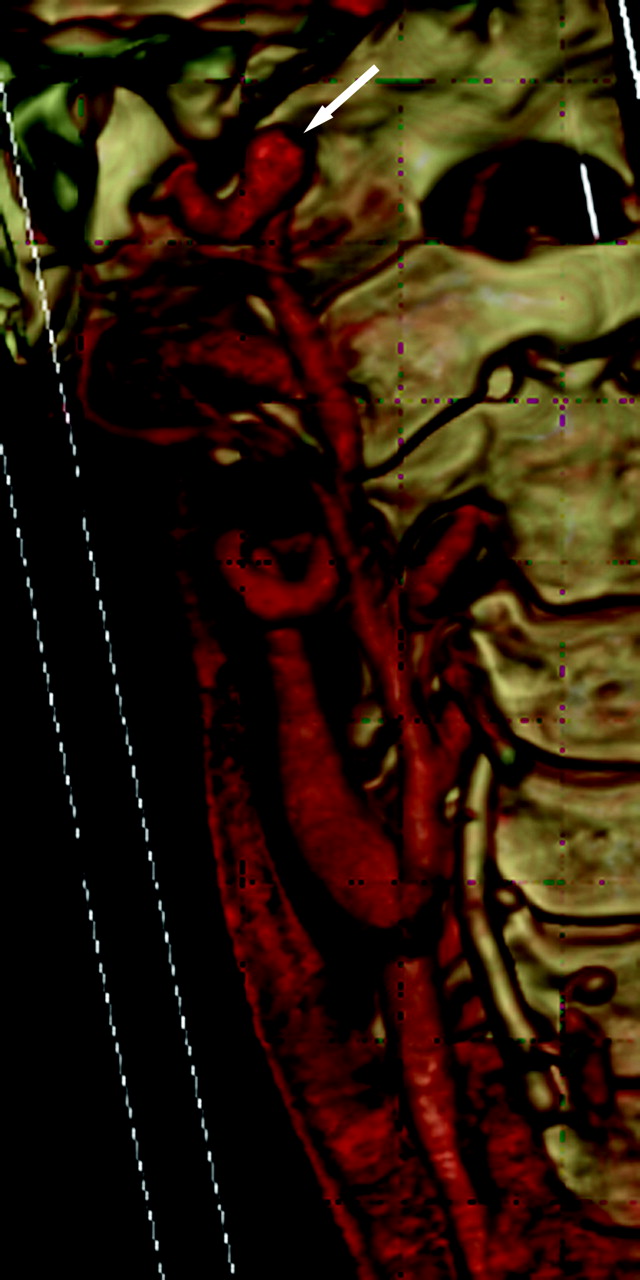
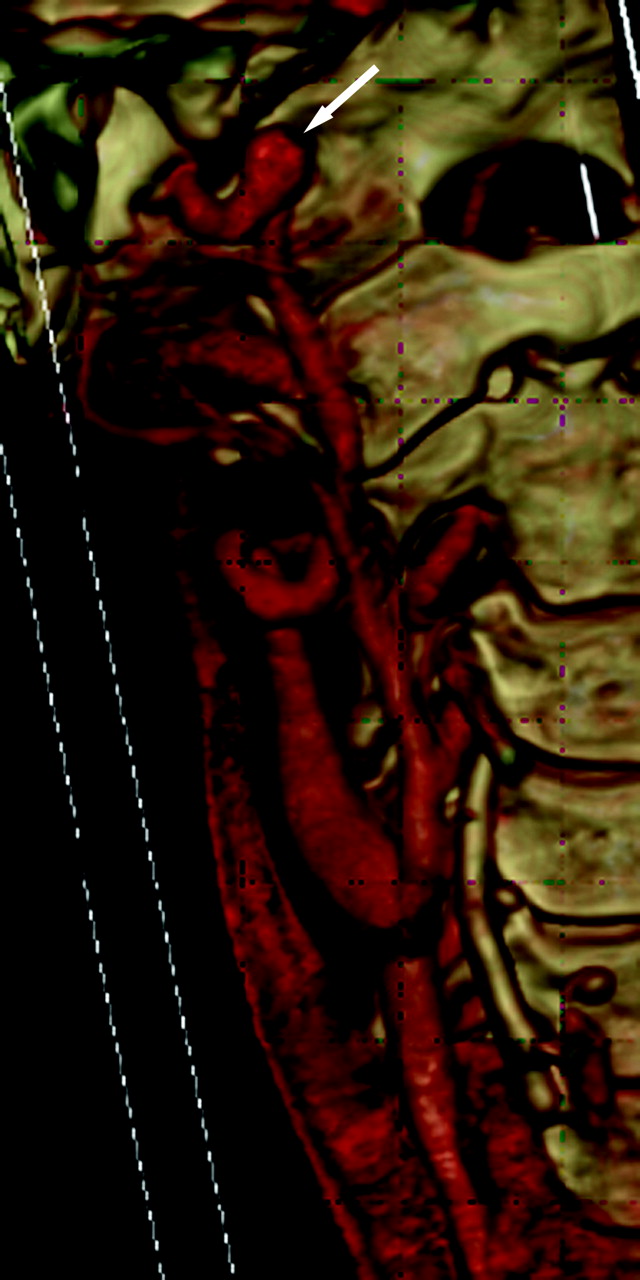
A 41-year-old woman with a family history of aneurysms and positive genetic testing for LDS type 1 had tortuous bilateral ICAs and a fusiform 8-mm aneurysm distal to the right ICA (arrow).

A 41-year-old man and his son had positive genetic testing for LDS type 1 after he had intracranial hemorrhage as a complication of dissection and some connective tissue features of LDS. Focal vertebral artery dissection is demonstrated on sagittal (A, arrow) and axial (B) views.
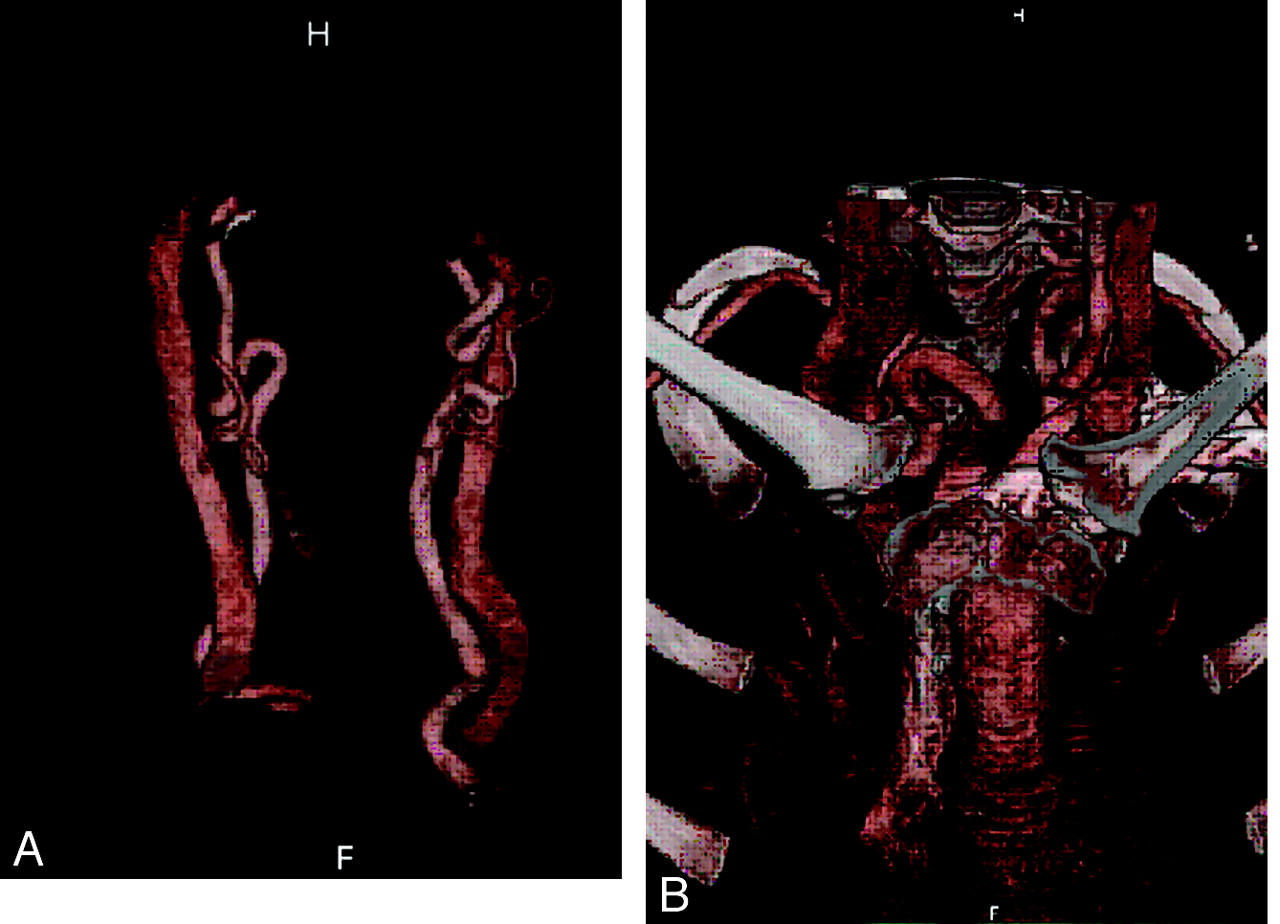
An 8-year-old genetically confirmed patient with significant arterial tortuosity is demonstrated in carotid (A) and supra-aortic branches (B).
Twelve (48%) of 25 patients had craniosynostosis: 8 had premature fusion of the sagittal suture (dolichocephaly), 2 had premature fusion of the coronal suture (brachycephaly), 1 had premature fusion of the metopic suture (trigonocephaly), and 1 had premature fusion of both the sagittal and metopic sutures.
Two patients (8%) had a Chiari 1 malformation, 1 of whom required surgery for posterior fossa decompression. Although none of the patients with Chiari 1 malformations had hydrocephalus, there were 3 others (12%) with mild-to-moderate communicating hydrocephalus, the cause of which was not known. Such idiopathic communicating hydrocephalus has been associated with this connective tissue disorder. Two (8%) patients had nonobstructing arachnoid cysts in the posterior fossa ranging from 1 to 2 cm without hydrocephalus.
Spine
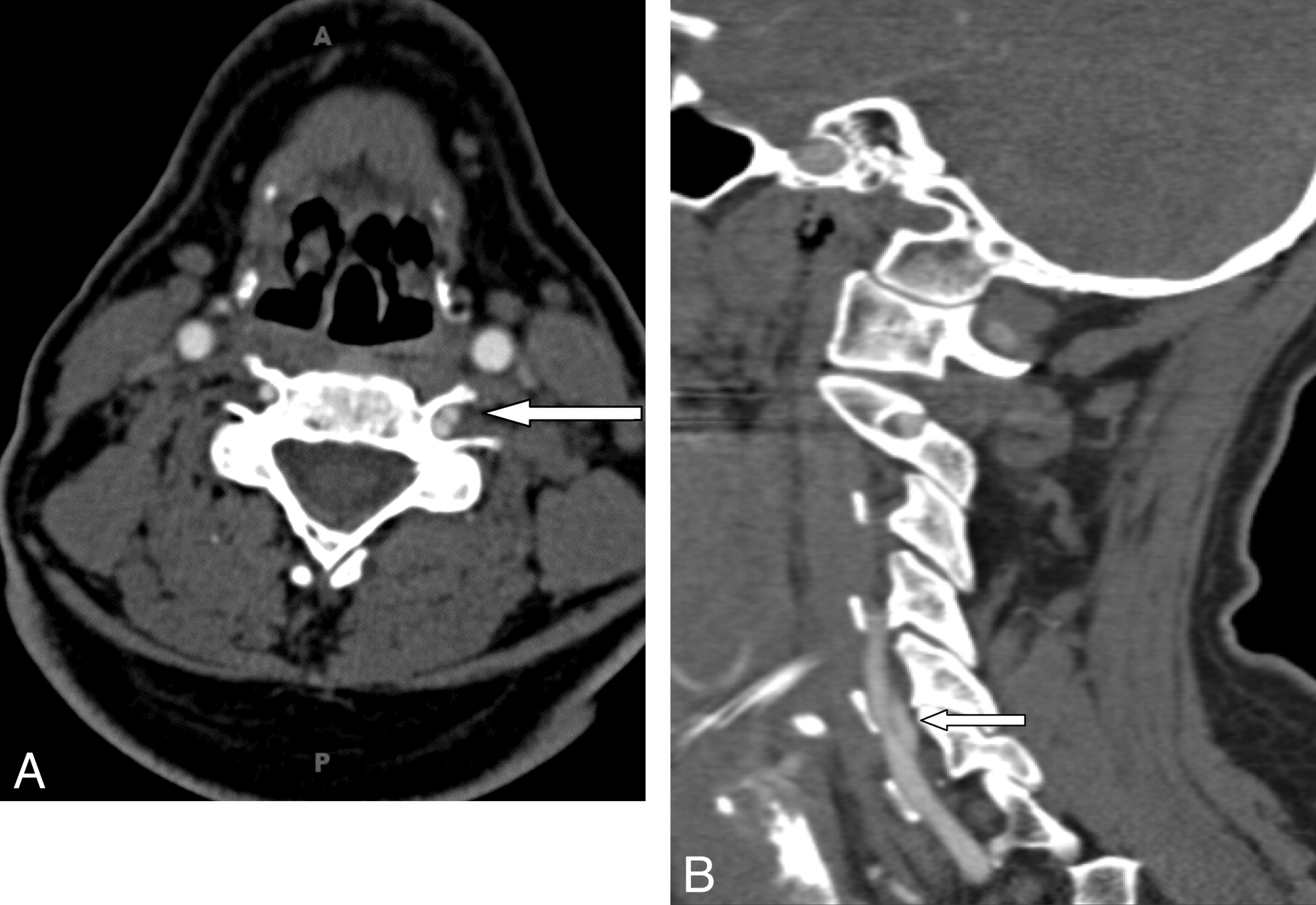
Thirteen (52%) of 25 patients had spinal scoliosis on review of the plain films. The scoliosis was thoracolumbar and mild, with angles of <15°. One patient had significant scoliosis >30°, which required surgery. Other alignment abnormalities included 6 (24%) patients with mild spine instability on flexion/extension views, 4 of which were cervical and 2, lumbar spondylolisthesis (Fig 4). Four (16%) patients had dural ectasias, which were lumbosacral and mild (Fig 5).

A 29-year-old male patient with positive genetic testing and dural ectasia noted on CT of the abdomen and pelvis and other connective tissue manifestations like bilateral shoulder.

An 11-year-old girl with positive genetic testing and other connective tissue manifestations demonstrates spine instability at both C1 and C2 (note atlantoaxial distance variation) and at C2 through C3 with flexion (A) and extension (B) views.
Clinical Manifestations
Ocular.
Twenty (80%) patients had hypertelorism (6 subtle and 14 marked), which was factored into the craniofacial index (CFI). On clinical examination, the globes of 11 (44%) patients showed a blue sclera and 7 (28%) patients had exotropia. One patient had a retinal detachment diagnosed on funduscopy and not detected on CT.
Facial.
Fourteen (56%) patients had a bifid uvula, 5 had a broad uvula, 4 had a midline raphe, and 2 had a normal uvula. These findings were based on clinical examinations documented in notes and also were factored into craniofacial indices. The palates were abnormal in nearly all patients: 6 (24%) had a cleft palate, 1 had a submucous cleft at birth, 20 (80%) had a high arched palate, and 1 had a narrow palate. The facial bone evaluation both by clinical and CT imaging studies revealed 14 (56%) patients with micrognathia, 12 (48%) with retrognathia, and 15 (60%) with malar hypoplasia.
CFI.
The CFI ranged from 1 (2 patients) to 11 (3 patients) (mean, 5.8 ± 2.9). Forty percent had a CFI of 6 or 8. The 2 patients with a CFI of 2 had family members with characteristic facial features of the syndrome and other vascular manifestations with arterial tortuosity. They also had positive genetic testing for the syndrome.
Discussion
Types
LDS is characterized by the triad of arterial tortuosity and aneurysms, hypertelorism, and bifid uvula or cleft palate.1,2 There are 2 types of LDS as described in the OMIM criteria:
-
1) LDS type 1: Craniofacial involvement consisting of cleft palate, craniosynostosis, or hypertelorism.
-
2) LDS type 2: Isolated bifid uvula and no other craniofacial features, but with at least 2 of the findings associated with vascular Ehlers-Danlos syndrome (EDS) (visceral rupture, easy bruising, wide and atrophic scars, joint laxity and translucent skin, velvety skin, or both).
These patients had previously received a provisional diagnosis of vascular EDS after evaluation by a medical geneticist, but the diagnosis of EDS was subsequently ruled out by negative studies of type III collagen biosynthesis. All patients had positive TGFBR1 and TGFBR2 mutations.
All our patients were classified as LDS type 1.
Molecular Genetics
TGFBR signaling has a prominent role in vascular and craniofacial development, and perturbations of TGFBR signaling occur in many human phenotypes, including craniosynostosis, cleft palate, arterial aneurysms, congenital heart disease, and mental retardation.1–5
Histologic analysis in patients with LDS with mutations in TGFBR2 shows loss of elastin content and disarrayed elastic fibers in the aortic media similar to those in patients with Marfan syndrome. Structural analysis shows loss of intimate spatial association between elastin deposits and vascular smooth muscle cells and a marked excess of aortic wall collagen. These characteristics are observed in young children and, in the absence of inflammation, suggest a severe defect in elastogenesis rather than secondary elastic fiber destruction.5
Neuroradiologic Features
Previous limited descriptions of LDS have mentioned possible neuroradiologic features including those affecting the brain and skull, such as craniosynostosis, intracranial arterial tortuosity, aneurysms, dissection and stroke, Chiari 1 malformation, and hydrocephalus. Spine manifestations include scoliosis, spine instability, and dural ectasia. Head and neck features, such as cleft palate, high arched palate, hypertelorism, and cervical vascular tortuosity and/or aneurysms, may be present. There are other craniofacial anomalies not visible on imaging, like blue sclera, bifid uvula, micrognathia, retrognathia, exotropia, and malar hypoplasia, which have been reported in LDS2 and which were gleaned from clinical notes on our patients. In addition, neurocognitive features, like developmental delay, headaches, lethargy etc, have also been noted.2
The non-neuroradiologic features include pectus excavatum or carinatum, hypermobility and dolichostenomelia, camptodactyly (contractures of fingers and toes), congenital heart diseases like patent ductus arteriosus and atrial septal defects, translucency of the skin with velvety texture, aortic root dilation, and aggressive aneurysms and dissections throughout the body including the coronary arteries.6,7
Our study reports on CNS and head and neck manifestations of LDS in 25 well-documented genetically positive patients with LDS type I (Table). The most common imaging findings in this study were vascular tortuosity (100%), scoliosis (52%), and craniosynostosis (48%). Aneurysms in the carotid and vertebrobasilar systems (32%) and the high rate of dissections (12%) may require urgent treatment to prevent more catastrophic complications. Neurointerventional consultations may lead to preventive measures being addressed to forestall strokes and intracranial/subarachnoid hemorrhages. Spine instability in 24% of patients also poses a potential threat to the CNS axis, which may need to be promptly addressed. Hydrocephalus (12%) was not associated with Chiari malformation and is due to underlying connective tissue abnormality.
Comparison of the current study and a previously published report* on CNS and head and neck findings in LDS type I
Previous studies were focused on the systemic manifestations of the LDS. On review of the previously published articles by Loeys et al,1,2,4 our results were concordant for similarly described findings as seen in the Table. The only significant discrepancies were that all our patients had tortuosity of the head and neck vessels and 84% of patients in the previous series had systemic arterial tortuosity.1,2,4 Fifty-two percent of patients in their series had arterial aneurysms somewhere in the body, whereas we reported a 32% incidence of aneurysms of the intra- and extracranial carotid and/or vertebral vessels and a 12% incidence of neurovascular dissections and/or pseudoaneurysms. Also in our series, only 2 of 25 patients imaged had a Chiari 1 malformation whereas 4 of 40 patients in the Loeys et al1 series had a Chiari 1 malformation.
Differential Diagnosis of LDS
The differential diagnosis of LDS includes the following: 1) atypical Marfan syndrome, 2) familial thoracic aortic aneurysm and dissection syndrome, 3) vascular EDS, and 4) Shprintzen-Goldberg craniosynostosis. The genetic tests described above and the clinical OMIM and neuroimaging features described herein distinguish LDS from these other entities.
Marfan syndrome is caused by mutations in the fibrillin-1 (FBN1) gene located on chromosome 15q21, which encodes fibrillin-1, which is essential for formation of elastic fibers that are abundant in the aorta, ligaments, ciliary zonules of the eye, and other connective tissues. Another mechanism is that fibrillin binds to TGFBR and inactivates it; reduced fibrillin increases TGFBR, which causes damage. There are skeletal, ocular, and cardiovascular manifestations as well as dural ectasia and pulmonary and skin findings but no neurocognitive findings in patients with Marfan syndrome. Also most patients with Marfan syndrome have ectopia lentis, which is not associated with LDS.2,3
Familial thoracic aortic aneurysm and dissection syndrome maps to 3p25-p24, caused by mutations in the TGFBR2 gene. Multiple thoracic aneurysms and dissection are a feature of this syndrome, but no ocular or skeletal features of Marfan syndrome are associated.8,9
Vascular EDS (type 4) is autosomal dominant in type 3 collagen. It is characterized by delicate fragile skin with a tendency for organs to rupture or develop aneurysms, life-threatening complications in pregnancy, characteristic facial appearance (thin lips and philtrum, small chin, thin nose, large eyes), acrogeria, hypermobility of small joints, etc, and overlap with features of type 2 LDS.
Shprintzen-Goldberg craniosynostosis syndrome is a marfanoid craniosynostosis syndrome, with ocular hypertelorism, downward slanted palpebral fissures, a high and narrow palate, mandibular micrognathia, arachnodactyly, camptodactyly, inguinal and umbilical hernias, hypotonia, mental retardation, and other skeletal and connective tissue defects. Shprintzen-Goldberg craniosynostosis syndrome is not associated with cleft palate, arterial tortuosity, or risk of aneurysm or dissection other than of the aortic root.10
Conclusions
Widespread neuroradiologic manifestations were encountered in our series of 25 patients with LDS, all of whom had arterial tortuosity; 3 had dissections and 8 had intra- and extracranial aneurysms. Most of the patients in this series were young and, therefore, will require serial CTA monitoring for development of intra-and extracranial dissections and aneurysms and appropriate interventions to forestall neurologic complications.
Patients should be advised of and evaluated for other neuroradiologic manifestations of the disease that are treatable like spine instability, scoliosis, Chiari 1 malformation, craniosynostosis, cleft palate, etc.
Given its substantial overlap with Marfan syndrome and vascular EDS, including the extensive involvement of the aorta, skeleton, and dura, it is important to assess the craniofacial and neuroradiologic manifestations that are not typical of these 2 other syndromes.
LDS is an aggressive aortic and distal artery aneurysm syndrome with a propensity toward rupture and dissection at a younger age, and early recognition of the phenotype and meticulous surveillance of the vascular tree are warranted for optimal management.
References
- Received January 23, 2009.
- Accepted after revision April 1, 2009.
- Copyright © American Society of Neuroradiology
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Infant with Loeys-Dietz syndrome treated for febrile status epilepticus with COVID-19 infection: first reported case of febrile status epilepticus and focal seizures in a patient with Loeys-Dietz syndrome and review of literature
- Value of Emergent Neurovascular Imaging for "Seat Belt Injury": A Multi-institutional Study
- Intracranial Arterial Tortuosity in Marfan Syndrome and Loeys-Dietz Syndrome: Tortuosity Index Evaluation Is Useful in the Differential Diagnosis
- Prevalence of Intracranial Aneurysms in Patients with Connective Tissue Diseases: A Retrospective Study
- Cerebral arterial angioplasty in a patient with Loeys-Dietz syndrome
- Cerebral arterial angioplasty in a patient with Loeys-Dietz syndrome
- Endovascular treatment of intracranial aneurysms in Loeys-Dietz syndrome
- Aggressive Cardiovascular Phenotype of Aneurysms-Osteoarthritis Syndrome Caused by Pathogenic SMAD3 Variants
- Increased Vertebral Artery Tortuosity Index Is Associated With Adverse Outcomes in Children and Young Adults With Connective Tissue Disorders