
Abstract
Summary: We describe two original cases of internal carotid artery dysgenesis associated with a malformative spectrum, which includes transsphenoidal encephalocele, optic nerve coloboma, hypopituitarism, and hypertelorism. Cephalic neural crest cells migrate to various regions in the head and neck where they contribute to the development of structures as diverse as the anterior skull base, the walls of the craniofacial arteries, the forebrain, and the face. Data suggest that the link between these rare malformations is abnormal neural crest development.
A malformative spectrum has been described in the literature, which associates, to various degrees, transsphenoidal encephalocele, facial dysmorphia, dysgenesis of the corpus callosum, hypopituitarism, and optic disk anomalies. To our knowledge, dysgenesis of the internal carotid artery (ICA) has never been reported in association with this malformative spectrum. We describe two original cases of ICA dysgenesis associated with transsphenoidal encephalocele, optic nerve coloboma, hypopituitarism, and hypertelorism. The classic theory attributes transsphenoidal encephalocele to a faulty separation of neurectoderm from the surface ectoderm during neural tube formation that thereby prevents mesodermal tissue from interposing between the two germ layers. We propose a new explanation for this malformative spectrum in light of our new findings and of the advances that have been made in the field of head and neck embryology during the last decade, all of which suggest the involvement of aberrant neural crest cell development.
Case Reports
Case 1
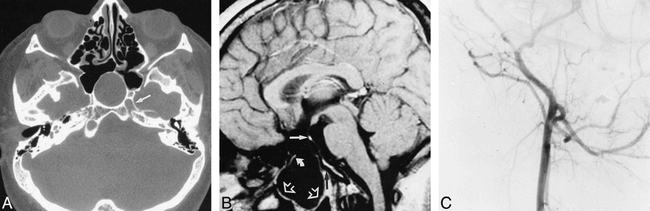
A 23-year-old man presented with progressive headaches. His clinical history revealed a panhypopituitarism requiring hormonal substitution therapy and congenital microphthalmia with cataract of the right eye. A physical examination revealed short stature, hypertelorism, and normal neurologic status. CT of the head revealed a mass of liquid density extending downward from the suprasellar cistern, through a large bone defect in the floor of the sella turcica and the sphenoidal sinus, into the rhinopharynx (Fig 1A). It also showed an absence of the right carotid canal (Fig 1A) and the presence of an optic nerve coloboma of the right eye. MR imaging of the head showed a transsphenoidal encephalocele, an optic chiasm that appeared elongated and atrophied, and an ectopic neurohypophysis with no identifiable adenohypophysis (Fig 1B). All other brain structures were normal. Cerebral angiography revealed segmental dysgenesis of the right ICA with filling of its supraclinoid portion through the posterior communicating artery, providing a normal middle cerebral artery (Fig 1C). The precommunicating segment of the right anterior cerebral artery was also agenetic. No other vascular anomalies were noted, and, in particular, no intracranial aneurysms were observed.

23-year-old man with progressive headaches.
A, CT scan obtained through the midcranial skull base in the axial plane shows a transsphenoidal encephalocele (black arrows) and complete absence of the right carotid canal. The left carotid canal can be seen (white arrow).
B, Cerebral MR imaging. Sagittal spin-echo T1-weighted sequence, 480/14/4 (TR/TE/excitations), shows a transsphenoidal encephalocele (open arrows). The bone defect involves the basipresphenoid, leaving intact the basipostsphenoid (black arrow). Atrophied optic chiasm pulled down into the bone defect can be seen (curved arrow). Unidentifiable adenohypophysis with ectopic neurohypophysis are shown (straight white arrow).
C, Angiogram of the right common carotid artery. Lateral view shows filling of the right external carotid artery alone.
Case 2
A 37-year-old man presented with progressive bitemporal visual field defect. His clinical history revealed a panhypopituitarism treated since childhood by hormonal replacement therapy and long-standing amblyopia of the left eye caused by an optic nerve coloboma. A physical examination revealed hypertelorism, obesity, and normal neurologic status. Spiral CT of the skull base revealed a mass of liquid density extending downward from the suprasellar cistern, through a large bone defect involving the rostral half of the sphenoid bone, into the rhinopharynx. It also revealed a hypoplastic right carotid canal and confirmed the coloboma of the left optic nerve (Fig 2A). MR imaging showed a transsphenoidal encephalocele, an optic chiasm that appeared elongated and atrophied, and no identifiable adenohypophysis, with a normally positioned neurohypophysis. All other brain structures were normal. MR angiography of the cranial arteries showed a right ICA with an extremely thin flow signal, which disappeared at the distal segment (Fig 2, B and C). A normal right middle cerebral artery was seen as well as a long right anterior cerebral artery, which was pulled down into the bone defect (Fig 2C). The left ICA appeared large and tortuous but provided normal cerebral branches (Fig 2C). No cerebral aneurysms were found.

37-year-old man with progressive bitemporal visual field defect.
A, CT scan of the midcranial skull base in the axial plane shows a transsphenoidal encephalocele (white arrows) and a narrow right carotid canal (black arrow).
B and C, MR angiograms obtained using the 3-D time-of-flight technique with 40/7/1, flip angle of 15°, 64 sections, and 1.2-mm section thickness.
B, Axial source image shows the narrow diameter of the right ICA within the carotid canal (arrow).
C, Maximum-intensity projection reconstruction in the anteroposterior view shows signal loss of the right distal ICA (curved arrow). The large left ICA (straight solid arrow) and anterior cerebral artery (open arrow) caused by the collateral blood supply to the right middle cerebral artery and to the right anterior cerebral artery provided by the left carotid system via the anterior communicating artery can be seen.
Discussion
Cephaloceles are congenital skull and dural defects with extracranial herniation of intracranial structures. Those involving the skull base are rare, with an estimated prevalence of one in 35,000 births (1). The transsphenoidal encephalocele is the least frequent type, representing 5% of all basal encephaloceles (1). One of the earliest and most widely accepted theories of basal encephalocele formation is the “adhesive theory,” published in 1827 by Geoffroy Saint-Hilaire (as cited by Smith et al [1]). This theory attributes the bone defect to a faulty separation of neurectoderm from the surface ectoderm during neural tube formation, thereby preventing mesodermal tissue, which is to form bone, from interposing between the two germ layers. None of the theories that have been advanced, however, can account for all of the associated malformations described with transsphenoidal encephaloceles, particularly dysgenesis of the ICA, which is reported herein.
Our understanding of skull base development has greatly advanced during the last decade, in large part because of the work of Couly et al (2), who provided an elegant illustration of the embryonic origin of all of the bones that compose the avian skull, using a quail-chick chimera technique. For the first time, they reported a dual origin of the sella turcica, which divides the skull base into a rostral half of neural crest origin and a caudal half of mesodermal origin (2). These authors found that the ethmoid, the basipresphenoid, the rostrum of the parasphenoid, and the pterygoid derive from neural crest cells whereas the basipostsphenoid, the alisphenoid, the basioccipital, and the exo-occipital bones derive from mesoderm (2).
In humans, migration of cephalic neural crest cells begins at approximately the fourth week of development. These cells form a series of condensations collectively known as the desmocranium, which represents the earliest evidence of skull formation (3). These condensations go on to form multiple chondrification centers, beginning at approximately the seventh week of development, which eventually fuse to form the chondrobasicranium between the developing brain and foregut (3). The fusion of the embryonic basicranial cartilages occurs around preexisting structures to form the skull base foramina, giving way to the blood vessels and cranial nerves of the skull base (3). If these blood vessels are of small caliber or are agenetic, as in our two cases, then their skull base foramina will likewise be narrow or agenetic (4). The sphenoid body contains two groups of endochondral ossification centers; the basipresphenoid centers, which are rostral, and the basipostsphenoid centers, which are caudal (5). At birth, the basipresphenoid contains the paired anterior accessory and main centers and the unpaired corporal middle center. Shortly after birth, the main centers join in the midline to form the border of the tuberculum sellae posteriorly and the anterior accessory centers do likewise to form the sulcus chiasmticus. Both of these structures lie above the corporal middle centers. The basipostsphenoid is composed of a pair of medial and lateral ossification centers. The paired medial centers completely fuse in the midline within 3 months after birth. Fusion between each medial center and the ipsilateral lateral center is usually completed by 6 months. The resulting structures lead to the formation of the caudal portion of the sellar floor in the midline and the dorsum sellae posteriorly. The intersphenoidal synchondrose, at the junction of the basipresphenoid and basipostsphenoid, undergoes complete closure by 3 months of age (5).
Considering the comparative anatomy and embryology of the skull base in the avian model, it is likely that the cephalic neural crest cells play a major role in the development of the rostral skull base in humans as well. In a review of the literature on the neuroradiologic appearance of cephaloceles, including 19 cases of transsphenoidal encephaloceles, the dorsum sellae was always found to be normal with the bone defect involving the rostral half of the sellar floor (6). In both of the cases reported above, the caudal limit of the bone defect is located at the midpoint of the sella turcica. In further support of the role of aberrant neural crest development in the constitution of basal encephaloceles, Goldhammer and Smith (7) reported a case of combined transsphenoidal and transethmoidal encephaloceles in a patient with Goldenhar's syndrome, considered to be a neurocristopathy involving neural crest derivatives of the first and second branchial arches.
Coloboma of the optic nerve head, which results from faulty closure of the fetal cleft during organogenesis, is one of a number of optic nerve anomalies that have been reported in association with basal encephaloceles (7). Hypothalamic-pituitary dysfunction is also commonly associated with a variety of prechordal skull base malformations, including transsphenoidal encephaloceles (8). It has been speculated that both of these anomalies associated with basal encephaloceles develop as a result of traction, which draws these structures into the bone defect. On the other hand, a number of studies have shown the involvement of developmental control genes, expressed in structures that derive from the anterior neural plate and that are implicated in the genesis of the ventral forebrain, including the optic recess, hypothalamus, and optic vesicles (9). It has also been shown that under the inductive influence of the cephalic neural crest cells that have migrated to the future prechordal skull base, the overlying neural tube is transformed, first, into the prosencephalon and then its differentiation into the diencephalon and telencephalon occurs (10). In the absence of neural crest cell migration to the future prechordal skull base region, the overlying diencephalic neural tube may not undergo appropriate induction during organogenesis of the optic nerves and hypothalamic-pituitary axis. The patient in case 1 presents an ectopic neurohypophysis, which may also be a sign of abnormal diencephalic development.
ICA dysgenesis is the congenital absence of either a portion or all of the blood vessel. Its actual prevalence is unknown because it is often clinically silent. The mechanism involved in dysgenesis of the ICA remains controversial. It may result from either regression at a late stage after an initial period of normal development or, more probably, from an early interruption of development. In a review of the patterns of cellular movement and tissue assembly during craniofacial development, Noden (11) cited several studies that have shown that the smooth muscle cells and connective tissue forming the walls of the aortic arches and most of the craniofacial arteries are of cephalic neural crest origin. The neural crest cells first form concentric layers around the vascular endothelium and differentiate to form the specific layers of the arterial wall.
Legius et al (12) reported a case of ICA dysgenesis with contralateral Goldenhar's syndrome. These authors suggest that both anomalies are the result of the same problem in migration of or interaction with cells derived from the cephalic neural crest. ICA dysgenesis has also been reported in several cases of a complex neurocutaneous syndrome called “cutaneous hemangioma-vascular complex syndrome,” which includes extensive cutaneous cervicofacial hemangiomas associated with anomalies of the cervical cranial arteries, cerebrovascular arteries, cerebellum, and, less frequently, the cerebral hemispheres and aortic arch (13).
Although no other cases of the transsphenoidal encephalocele spectrum malformation have been described as occurring in association with ICA dysgenesis, one case was reported of a 6-year-old boy with transsphenoidal encephalocele, coloboma, and hypopituitary dwarfism associated with stenosis of the right ICA and abnormal fine vessels in the basal ganglia similar to basal moyamoya network (14). It is not clear, however, from the description of the angiogram, whether the ICA stenosis is acquired or congenital, and no mention is made of the size of the carotid canal.
In both of our cases, CT of the skull base showed either an absence or hypoplasia of the right carotid canal, indicating the congenital nature of ICA anomalies. In case 1, the results of conventional angiography are consistent with the diagnosis of complete ICA dysgenesis. In case 2, in the absence of conventional angiography, it is difficult to assess the type of ICA dysgenesis. The imaging data observed in this case may correspond either to hypoplasia or distal segmental dysgenesis of the ICA. As noted by Lasjaunias and Berenstein (15), segmental dysgenesis distal to the dorsal ophthalmic artery gives, as in our case, a proximal hypogenetic ICA and collateral recruitment of the circle of Willis.
Conclusion
This original report of ICA dysgenesis associated with the transsphenoidal encephalocele spectrum malformation throws new light on the pathophysiology involved in this malformation. Several indicators point to the critical role of the cephalic neural crest in this malformative spectrum, providing a comprehensive pathophysiologic explanation. We suggest that this transsphenoidal encephalocele spectrum malformation associated with ICA dysgenesis constitutes a neurocristopathy.
Footnotes
↵1 Presented in part at the American Society of Neuroradiology 36th Annual Meeting in Philadelphia, PA.
2 Address reprint requests to Jean François Meder, Department of Neuroradiology, Centre Hospitalier Sainte-Anne, 1 rue Cabanis, 75674 Paris, France.
References
- Received June 20, 1998.
- Copyright © American Society of Neuroradiology




{kind=link}
{kind=link}