
Abstract
Summary: We present the MR imaging findings in an autopsy-proven case of selective neuronal necrosis involving the entire left cerebral hemispheric cortex, left thalamus, and contralateral cerebellum following a period of status epilepticus. Imaging findings include diffusion abnormality on diffusion-weighted images and increased intensity on T2-weighted images in the above-mentioned regions of the brain.
Status epilepticus is defined as a condition characterized by an epileptic seizure that is sufficiently prolonged or repeated at sufficiently brief intervals, which produces an unvarying and enduring epileptic condition (1). Status epilepticus is consistently associated with widespread neuronal necrosis in vulnerable regions of the brain, evidenced both by neuropathologic studies in humans and experimental studies in animal models (1, 2). The literature concerning imaging findings in human status epilepticus mainly comprises a limited number of case reports (3–7). The imaging findings in these reports, which were obtained by use of various imaging techniques, are inconsistent with each other in most aspects, and most importantly, lack a correlative tissue diagnosis. Here we report the MR imaging findings in a case of autopsy-proven massive neuronal necrosis and multiorgan failure secondary to status epilepticus.
Case Report
A 24-year-old man with a history of generalized tonic-clonic seizures presented with a prolonged, witnessed, generalized seizure at an outside hospital. He had experienced the first seizure 4 years prior to admision, followed by another generalized seizure 1 year later. MR imaging obtained after the second seizure had shown a small right hippocampal lesion thought to be dysembryoplastic neuroepithelial tumor or low-grade glioma. He had been placed on valproic acid, and with that medication had been seizure-free until the day of the admission. At admission, his blood valproic acid level was 50 μmol/L (therapeutic level, 350–700 μmol/L); his rectal temperature was 41.9°C, and he still had generalized tonic-clonic seizures. He continued to have right-sided focal motor seizures. Because he did not respond to benzodiazepines and valproic acid, he was intubated and anesthetized with profopol, which finally controlled his seizures approximately 24 hours after admission.
On the 2nd day of admission, he became oliguric and then anuric, with a serum creatinine level of 400 μmol/L (normal, 62–120 μmol/L). Because medical therapy of his acute renal failure failed to reestablish any urine output, early hemodialysis was instituted. His serum creatine phosphokinase level was 20 000 IU (normal, 38–174 IU). Concurrent abnormalities included disseminated intravascular coagulation evidenced by increased International Normalized Ratio, partial thromboplastin time, d-dimer level, and severely decreased platelet count.
On the 3rd day, he was still hyperpyrexic. His chest radiograph showed right upper lobe infiltrate. His liver function tests were elevated, as were his amylase and lactic dehydrogenase levels. He was hypoglycemic.
On the 4th day, the patient was transferred to our hospital. He was unconscious, intubated, hypotensive, anuric, hypoglycemic, slightly jaundiced, and febrile. Neurologically he had better function of the left extremities than the right, with an upgoing toe on the right and ability to withdraw the left upper extremity to pain. He attempted to open his left eye to deep pain. Glasgow Coma Scale score was 3. Lumbar puncture was unremarkable. He underwent cranial MR imaging. Diffusion-weighted images (Fig 1A and B) showed abnormal diffusion throughout the cortex of the left hemisphere, left thalamus, and the right cerebellar hemisphere. Only the cingulate gyrus and the temporal lobe were partially preserved in the left cerebral hemisphere. The involved hemispheric cortical tissue was thickened, and the cortical sulci were effaced on the left side. Increased signal intensity at the above-mentioned sites was present but less marked on the T2-weighted and fluid-attenuated inversion recovery (FLAIR) images (Fig 1C and D). Another notable finding was prominent vascular markings on the left hemisphere compared with the contralateral side (Fig 1C and E). The patient's known right temporal lesion in the amygdala and anterior hippocampus was again seen (Fig 1D). It was also noted that the white matter in the right temporal pole was less well developed and slightly brighter than its contralateral counterpart.
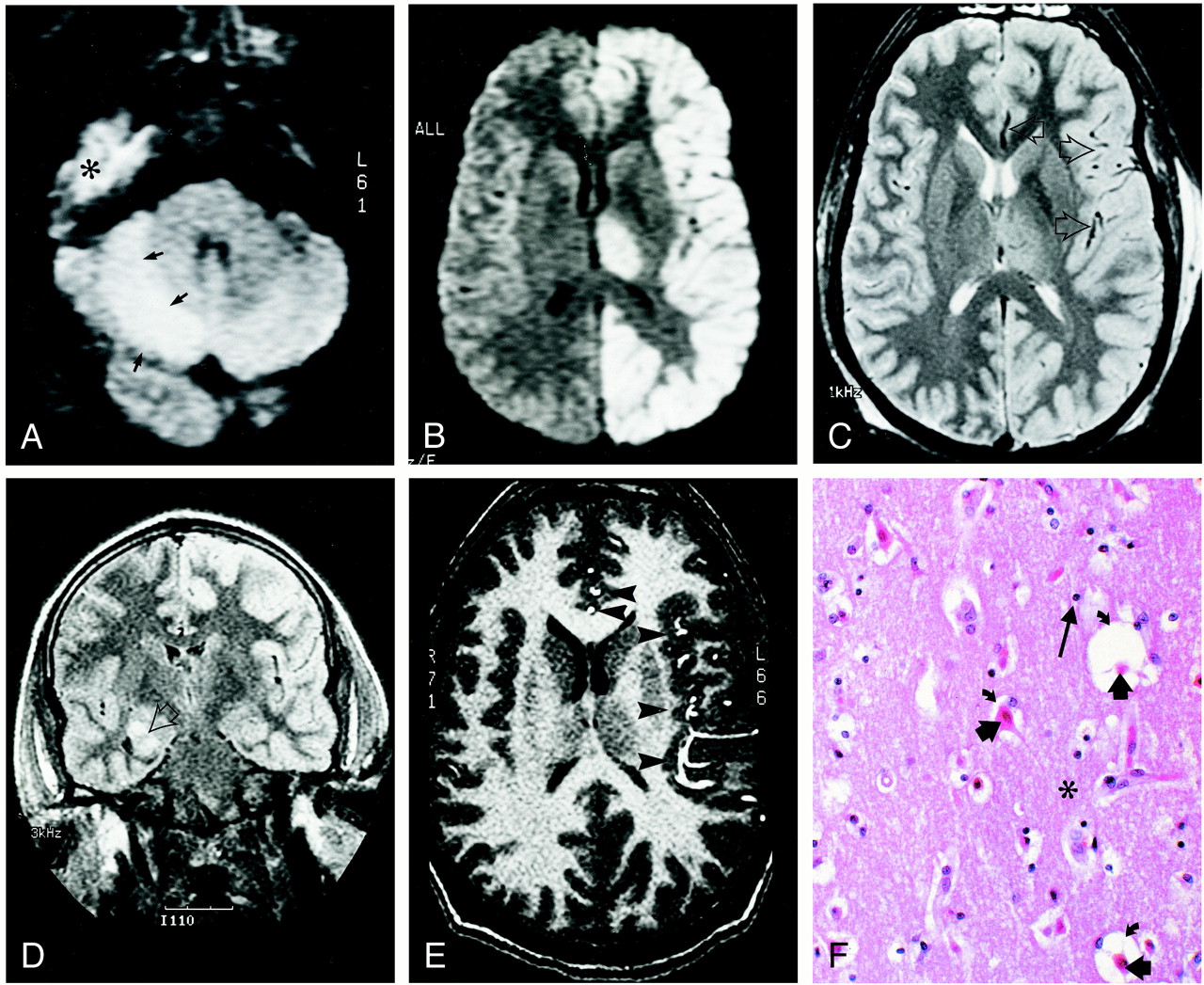
24-year-old man who experienced status epilepticus 4 days prior to imaging.
A and B, Diffusion-weighted trace images (9999/91.4 [TR/TE], B = 1000 s/mm2) through the levels of cerebellum (A) and thalami (B) show diffusion abnormality involving the right cerebellar cortex (arrows in A), the entire left hemispheric cortex, and the left thalamus (B). The high signal in the right middle fossa (indicated with * in A) is artifactual.
C, Spin-echo T2-weighted (2800/80/1 [TR/TE/excitations]) image through the same level as B shows mildly increased intensity in the diffusely thickened left hemispheric cortex and the left thalamus. Note the relative prominence of signal void in middle and anterior cerebral artery branches (arrows) on the left side.
D, Fluid-attenuated inversion recovery coronal image (9002/145/2200 [TR/TE/TI]) reveals the right hippocampal ganglioglioma (arrow). Note increased intensity in the left hemispheric cortex.
E, Flow-sensitive gradient-echo T1-weighted image (9.3/2.1/90 degrees [TR/TE/flip angle]). Prominent anterior and middle cerebral artery branches (arrowheads) over the left hemispheric cortex suggest enhanced arterial flow in the left hemisphere.
F, Histologic section through the left frontal cortex stained with hemotoxylin and eosin shows eosinophilic neurons (short, thick arrows). The surrounding neuropil (indicated with *) is edematous, as demonstrated by expanded perineural spaces (curved arrows), but there is no fragmentation of the neuropil, and glial cells (long arrow) showed no evidence of necrosis or apoptosis. These findings correspond to selective neuronal necrosis.
Over the course of the next 2 days, the patient went on to develop fulminant hepatic failure, pleural effusions, and diffuse air space infiltration in both lungs. Serologic tests for viral hepatitis and human immunodeficiency virus were negative. He expired on the 6th day. Autopsy of the body showed bilateral pleural effusions, pulmonary edema with areas of consolidation, and diffuse alveolar damage in the lungs. There was marked centrilobular zonal necrosis in the liver and multiple hyalin casts within renal tubules, consistent with acute hepatic and renal failure. Patchy subendocardial hemorrhage, scattered visceral petechiae in the pericardium, scleral hemorrhage, and hemorrhagic gastritis were other findings at general autopsy.
Macroscopic neuropathologic findings included softening of the cortex and the white matter of the left hemisphere, accompanied by cortical pallor. Light microscopy of sections throughout the left cerebral hemisphere showed abundant neurons with eosinophilic change in a transcortical distribution. The eosinophilic change was accompanied by marked cortical edema manifested by expanded perineural spaces (Fig 1F); however, the neuronal density was preserved, there was no fragmentation of the neuropil, and glial cells showed no evidence of necrosis or apoptosis. There appeared to be no correspondence between the relative intensities of edema and the extent of the eosinophilic change. Corresponding regions of the right cerebral hemisphere did not show any neuronal change. Both sides demonstrated abundant Alzheimer type II astrocytes throughout the cortex. Extensive neuronal eosinophilic cell change was also seen in the left hippocampus, affecting essentially all the neurons in the CA4, CA3, and CA1 sectors, sparing the CA2 sector. The change also spared the presubiculum and entorhinal cortex, and was present again in the basal temporal neocortex. The right hippocampus did not show any eosinophilic cell change. There was a tumoral lesion in the right amygdala, 0.7 cm in diameter, with histopathologic characteristics of ganglioglioma. The left thalamus showed extensive neuronal eosinophilic change. Alzheimer type II astrocytes were present in the thalami and basal ganglia on both sides. The right cerebellar neocortex showed loss of many Purkinje cells accompanied by proliferation of Bergman glia in reaction to Purkinje cell loss. Many of the remaining Purkinje cells and many neurons in the right cerebellar dentate nucleus showed eosinophilic cell change. Widespread Alzheimer type II astrocytes were observed in both cerebellar hemispheres. The above-mentioned findings were interpreted as recent, extensive, selective neuronal necrosis in the left cerebral neocortex and hippocampus, and the right cerebellum. The widespread finding of Alzheimer type II astrocytes was consistent with hepatic encephalopathy.
Discussion
Selective neuronal necrosis is a well-known consequence of status epilepticus. The most important factor linking seizure activity to selective neuronal death seems to be the entry of sodium and, more particularly, calcium into neurons. Overloading the mechanisms for intracellular sequestration of calcium and outward transport of calcium is associated with marked increases in the intracellular concentration of calcium that activates a wide range of enzymes with diverse secondary consequences. In status epilepticus, glutamate, aspartate, and acetylcholine are thought to play major roles as excitatory neurotransmitters, whereas gamma amino butyric acid may be the dominant inhibitory transmitter. The excitotoxic mechanisms are mediated by N-methyl-d-aspartate receptors. Activation of these receptors during the course of epileptic discharges plays a crucial role in the enhanced calcium entry and subsequent cell death (1).
The left cerebral cortex, thalamus, and the contralateral cerebellar cortex, which showed selective neuronal necrosis on autopsy, presented as high signal intensity on diffusion-weighted images in our case. An experimental study on rats has shown that diffusion-weighted imaging findings obtained after status epilepticus are similar to those seen in stroke (8). The authors produced prolonged complex partial seizures in rats by systemic administration of kainic acid, an agonist for glutamate receptor subtype. They imaged the rats with diffusion- and T2-weighted sequences from 3 hours to 9 days after injection of kainic acid. In the piriform cortex and amygdala, the T2-weighted MR signal intensity uniformly increased from 24 hours to 72 hours, and returned to normal by 9 days. In the same regions, the apparent diffusion coefficient (ADC) decreased from 3 hours to 48 hours and then increased. The authors suggested that the initial ADC decrease was associated with neuropil swelling, and the subsequent increase in ADC resulted from neuropil fragmentation and consequent loss of diffusion barriers. The increased signal on T2-weighted images was attributed to the increase in total amount of water in the tissue. In another study, 23Na MR imaging and proton diffusion-weighted imaging were used in rats that had kainic acid–induced seizures (9). Within 24 hours after kainic acid injection, MR-visible sodium levels increased in the amygdala and piriform cortex, whereas the ADC decreased. Seven days later, sodium increases persisted, whereas ADC returned to normal. It was concluded that these findings were consistent with the hypothesis that sequential seizures caused perturbation of membrane ion homeostasis and an increase in sodium influx, which eventually evolved into irreversible cellular edema with MR-visible intracellular sodium and decreased ADC. Return of normal ADC and persistent high sodium levels at 7 days was explained by the increase in the extracellular space and tissue necrosis.
The above-mentioned imaging findings suggest a temporal evolution in seizure-induced brain damage. Such a temporal evolution of neuronal damage was proved in rats (2). In this study, neuronal damage detected in certain brain regions immediately after 3-hour-long seizures was slight-to-moderate, whereas it became more pronounced after a 24-hour recovery period. The degree of damage remained the same in 24-hour and 72-hour recovery groups. The imaging findings from the previous human case reports suggest a time course starting with increased intensity in diffusion-weighted images (5, 7), and T2-weighted images (4–7) obtained during or within a few days of status epilepticus. The increased intensity returns to apparent normality during the following weeks or months (4–7), with focal brain atrophy replacing the previous lesions (6, 7).
Increased focal brain perfusion associated with epileptic seizures is well known and has been shown by MR imaging, MR angiography (7), and single-photon emission CT (SPECT) (3). Recently, studies recruiting xenon CT, SPECT, positron emission tomography, and functional MR imaging have provided a considerable amount of data concerning cerebral blood flow and metabolism alterations associated with human epilepsy. Various types of seizures lead to 25% to 133% increase in focal cerebral blood flow and 80% to 120% increase in both focal and generalized cerebral metabolic rate for glucose (10). Relative increases in metabolism can persist for 24 to 48 hours after partial seizures, whereas increases in blood flow last for a considerably shorter time likely because of relative uncoupling of blood flow and metabolism. Complex partial seizures appear to lead to more prolonged elevations than simple partial seizures. The increased left hemispheric blood flow observed on the 4th day in our patient may reflect persistence of ictal blood flow increase, or it may have been caused by ongoing subclinical seizures undetected on scalp electroencephalography.
The parts of the human brain most vulnerable to damage from status epilepticus include the CA1 and CA3 segments and hilus of the hippocampus, amygdala, piriform cortex, cerebellar cortex, thalamus, and cerebral cortex (1). Although individually the involved brain regions in our case have been described in the literature, the combination of ipsilateral hemispheric cortical involvement with contralateral cerebellar involvement (crossed cerebellar diaschisis) has been rarely reported (3, 4, 7). Inclusion of the ipsilateral thalamus in this combination has been encountered in some cases, though less frequently (4, 7). Seizure activity propagates along specific anatomic pathways in the brain, and the particular pathways responsible for triggering and propagating a seizure determine the nature of the seizure and the pattern of motor manifestations that accompany it. The thalamic nuclei are principal relay sites for both subcortical and cortical projection systems, and accordingly represent a critical link in the transfer of information between cortical and subcortical structures, and between the periphery and the cortex. It is therefore reasonable to expect thalamic nuclei to be capable of regulating seizure succeptibility and influencing seizure propagation. Although various thalamic nuclei participate in seizure propagation, the dorsomedial nucleus seems to have an important role and it is damaged during prolonged hemiconvulsions in humans (11). The cerebellum is thought to have an inhibitory function on epilepsy through release of the inhibitory transmitter, gamma amino butyric acid from the Purkinje cells, and Purkinje cell loss is a morphologic feature of severe epilepsy with many generalized convulsions. The loss of Purkinje cells might be explained by an increased demand for inhibition, which results in gamma amino butyric acid depletion and subsequent influx of calcium into neurons until toxic levels are reached (12). In cases of crossed cerebellar diaschisis, the ascending influence of the cerebellum seems to be transmitted mainly through the superior cerebellar peduncle, whose fibers principally reach the contralateral mesencephalon and diencephalon.
Our patient had a small right temporal lobe tumor showing MR imaging findings that may be common to ganglioglioma, low-grade glioma, and dysembryoplastic neuroepithelial tumor. Histopathologic diagnosis was ganglioglioma. A cerebral tumor is a common cause of seizures. Our patient had left hemispheric findings associated with status epilepticus, suggesting a left hemispheric focus. Various studies on epileptic patients with cerebral tumors have shown that 21% to 38% of these patients had a secondary epileptic focus arising from homotopic cortex contralateral to the tumor (13). The primary lesions in these patients give rise to satellite foci in distant, but synaptically related cerebral regions. These secondary foci eventually develop all the properties of the primary focus, including clinical seizures and establishment of secondary foci. The process of secondary epileptogenesis takes time and evolves through several stages. The pathways involved during secondary epileptogenesis include the callosal connections as well as the long intrahemispheric pathways such as uncinate, arcuate, and superior longitudinal fasciculi.
Status epilepticus has many systemic complications (1). Acute tubular necrosis and resultant renal failure is a consequence of prolonged and intense muscle contractions, with rhabdomyolysis and subsequent release of several proteins from the leaky muscle membrane. Large amounts of myoglobin can be detected in the urine and can cause acute tubular necrosis. Creatinine phosphokinase can be elevated 100-fold in blood. Hyperpyrexia and hyperkalemia are other consequences of increased and sustained muscle activity. Hyperpyrexia, in turn, may cause widespread endothelial damage that may induce disseminated intravascular coagulation (14). Hypoxia may occur during the apnea accompanying tonic phases of seizures. A combination of hypoxia and the liberation of large amounts of lactic acid during muscle contractions may result in profound metabolic acidosis. Hypoglycemia, early hypertension or late shock, and cardiac arrhythmias may complicate status epilepticus as a result of excessive autonomic stimulation (1). Although the exact mechanism remains unclear, status epilepticus may also cause fulminant hepatic failure (15). Almost all of the above-mentioned complications occurred in our patient, leading to his death. However, these systemic disturbances cannot explain the restricted regional distribution of selective neuronal necrosis, which must be construed as a direct consequence of seizure activity.
References
- Copyright © American Society of Neuroradiology
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- Status Epilepticus in Adults: A Review of Diagnosis and Treatment
- Isolated Reversible Thalamic Vasogenic Edema Following a Generalized Seizure
- Diffusion MRI abnormalities after prolonged febrile seizures with encephalopathy
- MRI volumetry of the thalamus in temporal, extratemporal, and idiopathic generalized epilepsy
- Visualization of Evolving Status Epilepticus with Diffusion and Perfusion MR Imaging