
Abstract
Summary: Although intracranial hemorrhage has frequently been found responsible for mortality in adult patients with Alagille syndrome (AGS), no specific underlying cause has been identified. We describe the case of severe subarachnoid hemorrhage in a 30-year-old woman harboring five intracranial aneurysms and multiple peripheral vascular anomalies. To evaluate a possible higher incidence of intracranial aneurysms, a study of the cerebral vasculature in all AGS patients by using noninvasive imaging techniques should be considered.
Alagille syndrome (AGS) is an autosomal dominant disease with incomplete penetrance and variable expression. It has a prevalence of 1/100,000 (1) and affects a variety of organ systems. When fully expressed, patients have intrahepatic bile duct paucity, chlolestasis, caradiovascular malformations such as pulmonary stenosis, opthalmologic abnormalities such as posterior embryotoxon, skeletal abnormalities such as butterfly-like vertebral arch defects, and characteristic facial dysplasia (2).
Case Report
A 30-year-old woman with AGS was found comatose after 3 days of severe headache. At the emergency department, she presented with a Glasgow coma scale of 6 (Hunt-Hess grade 5), and CT showed a subarachnoid hemorrhage (SAH; Fisher grade 4) and a right parietal subdural hematoma causing a midline shift of 0.5 cm.
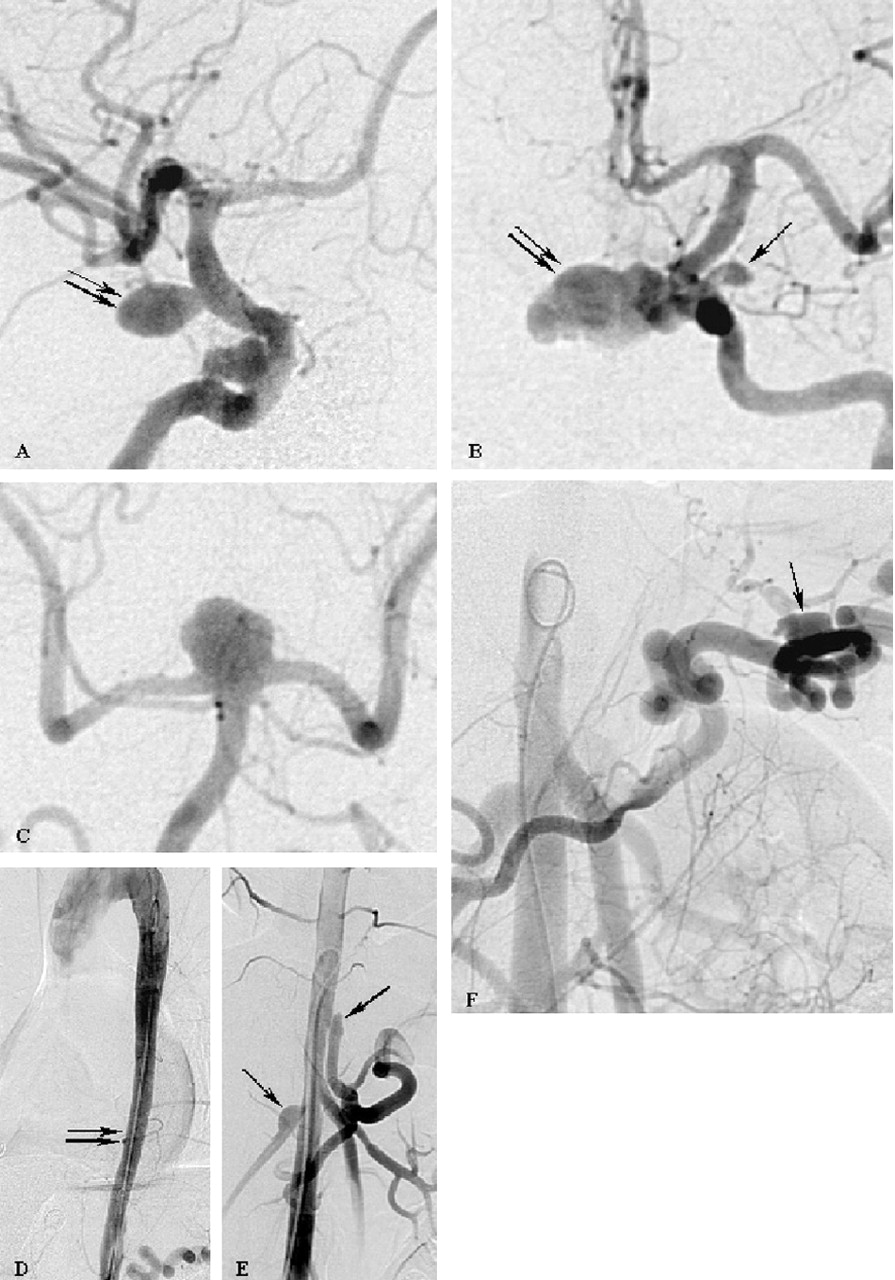
Cerebral angiography depicted a ruptured 8-mm aneurysm of the right supraclinoid internal carotid artery (Fig 1A). In addition, a small right extradural internal carotid artery aneurysm, two left internal carotid artery aneurysms (Fig 1B), and a basilar tip aneurysm (Fig 1C) were depicted. Furthermore, the abdominal angiogram showed aneurysms of the right renal artery, the celiac (trunk, the splenic artery, and a coarctation of the thoracic aorta (Fig 1D–F) without related clinical symptoms.

Cerebral (A-C) and abdominal (D-E) arteriograms demonstrate five intracranial aneurysms associated with multiple vascular abnormalities. Two saccular aneurysms of the right internal carotid artery are shown (A). The larger supraclinoid aneurysm (double arrow) caused the SAH and was treated by surgical clipping. The left internal carotid arteriogram (B) revealed a large infraclinoid aneurysm (double arrow) directed to the midline and an additional small aneurysm at the level of the origin of the ophthalmic artery (arrow). The left vertebral arteriogram demonstrated a broad-based aneurysm of the basilar tip (C).
In addition, a coarctation of the aorta (D, double arrows), aneurysmal formations at the origin of the right renal artery and of the celiac trunk (E, arrows), as well as an aneurysm of the splenic artery (F, arrow) are shown.
After surgical clipping of the right internal carotid artery, pathohistologic analysis of the arterial wall showed the typical structure of an aneurysm with an internal elastic lamina reduced to fragments and hypertrophied margins. The postoperative course was uneventful, and after rehabilitation the patient returned to normal life.
Exploration of the patient’s medical history revealed that AGS had been diagnosed in childhood. To treat the cholestasis caused by bilary duct dysplasia, the patient underwent a diversion procedure. Since then, the liver parameters were within the normal ranges. Even during her current hospitalization, the patient did not present any hepatic or cardiac dysfunctions and her laboratory parameters were within normal ranges. Echocardiography performed in her childhood revealed a valvular pulmonary insufficiency grade I, whereas regular cardiovascular consultations showed a normal cardiac function with a 3/6-holosystolic murmur over the second left intercostal space. As is typical for patients with AGS, she had facial dysplasia. She was able to work without restrictions and to take care of her family. Her brother died of a pulmonary valve insufficiency at the age of 35 years, and her mother underwent a cardiac valvular procedure.
Discussion
Most patients with AGS (86%) develop jaundice and chronic cholestasis before the age of 6 months (3). Many (70%) also have a cardiac murmur caused by peripheral pulmonary stenosis, which is frequently associated with other congenital heart abnormalities (4). Improvements in the therapeutic management of these abnormalities have improved the prognosis of these patients so that now most AGS patients have at least a 20-year life expectancy (1). Of those patients who live past childhood, stroke or intracranial hemorrhage (ICH) has been identified as a frequent cause of morbidity and mortality, with two recent series reporting such events in 13 of 92 and five of 43 patients with AGS, respectively (3, 4). Also, in these two series, the cause of death in four of 16 patients in one and one of 12 in the other was ICH. Unfortunately, in neither series, as well as in other smaller reports regarding ICH in patients with AGS, information provided regarding the specific cause of the hemorrhage is insufficient. Thus, despite the known occurrence of ICH in patients with AGS, there has not been, to our knowledge, a causal relationship established between ICH and a specific type of vascular lesion.
The report by Moreau et al (5) is, to our knowledge, unique in describing a saccular intracranial aneurysm in a patient with AGS. As many as 9% patients of some adult populations harbor a cerebral aneurysm. The incidence of SAH varies from 3.9–19.0 cases in 100,000, depending on geographic location. In the United States, the incidence of ruptured aneurysms is approximately 12 of 100,000, or 30,000, annual cases of aneurysmal SAH. Multiple cerebral aneurysms, as observed in our case, are seen in 15% of all patients with aneurysms in the normal population. Seven percent of these individuals have four or more aneurysms, which have been linked to a genetic abnormality affecting the elastin configuration. It is of interest that a genetic abnormality has been also described in AGS. The JAGGED1 gene, which has been shown to be mutated in AGS, encodes a transmembrane protein functioning as a ligand of Notch receptors (6). The Notch signaling pathway is connected to various developmental processes. It is known that mutations in the Notch signaling pathways have been associated with defects of vascular development in CADASIL, a cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (7).
Among various factors influencing the formation of berry aneurysms are genetic or possible genetic syndromes. Ehlers-Danlos syndrome, Marfan syndrome, pseudoxanthoma elasticum, Rendu-Osler-Weber syndrome, Klippel-Trenaunay-Weber syndrome, and type III collagen deficiency are known to be associated with cerebral aneurysms. A case of Moyamoya syndrome was reported by Woolfenden (8) and is further evidence of AGS patients’ inherent predisposition toward variform vasculopathy.
Whether the relatively high mortality of patients with AGS resulting from ICH is possibly related to previously unidentified SAHs caused by ruptured intracranial aneurysms remains questionable and can be answered only by more supporting data. For evaluation of a possible increased incidence of cerebral aneurysms in patients with AGS compared with that of the normal population, CT or MR angiography studies should be considered. The concurrence of multiple intracranial aneurysms and multiple extracranial vascular malformations in AGS has not yet been reported. Multiple peripheral vascular abnormalities, as found in our patient, are known and in agreement with other reports (9–11). Berard (10) found in his series a total obstruction or stenosis of one or both renal arteries in four patients that caused renovascular hypertension and a stenosis of the celiac trunk in one patient; however, an aneurysm of the splenic artery has not yet been described. Similarly, aortic coarctations have been reported in AGS (9, 10) and can be responsible for hypertension, intermittent claudication, or abdominal pain.
Conclusion
We describe a case of AGS in a patient with severe SAH in whom multiple and various cerebral and peripheral vascular lesion were detected. This case helps expand the existing knowledge of vascular abnormalities in AGS, emphasizing the predisposition toward vasculopathy in this genetic disorder. The limited data on the histologic nature of these vascular lesions in the literature, as well as the clinical importance of a ruptured berry aneurysm potentially causing fatal SAH, prompt more detailed evaluation of the cerebral vasculature in these patients by use of noninvasive imaging techniques such as CT or MR angiography.
References
- Received October 23, 2003.
- Accepted after revision December 24, 2004.
- Copyright © American Society of Neuroradiology





{kind=link}